Glycogen storage disease type I (GSDI) is an inherited genetic condition affecting the metabolism and its ability to continue normal functions. This is one of the common forms of glycogen storage disease and is also known as von Gierke’s disease. The body stores the excess amount of glucose as glycogen in the liver from the food we consume. This disease develops as a result of the enzyme responsible for the release of glucose from glycogen. The excess storage of glycogen in the body affects organs like the kidney, liver and small intestines. Hence, individuals with this condition have imbalanced glucose levels in the blood often developing hypoglycemia with symptoms such as hunger after eating and fatigue. The excess of glycogen storage creates liver swelling and enlarged kidneys as well. Infants with this disorder may have an excess of lactic acid and uric acid in their body. There are two subtypes GSI known as glycogen storage disease Ia and glycogen storage disease Ib. The incidence of this disease is 1 in 100,000 people, while GSDIa is more common accounting for 80% of the GSDI cases.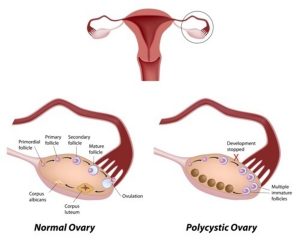
Symptoms
The symptoms become apparent at the age of 3 or 4 months old infants when they do not feed frequently as newborns. Infants suffering from hypoglycemia may result in seizures. Children with this disease indicate symptoms such as fatigue, growth failure, irritability, hunger, swollen abdomen and liver. Older children may have short stature with thin arms and legs. They are prone to have delayed puberty while the girls may develop polycystic ovaries. The affected individuals may develop gout, pulmonary hypertension and kidney disease.
Causes
The genetic mutations of two genes, G6PC and SLC37A4, are responsible for this disease. The gene G6PC is associated with the cause of GSDIa and SLC37A4 with GSDlb. The proteins created from these two genes assist in the breakdown of the sugar molecule called glucose 6-phosphate. This is essential in maintaining the stable blood sugar levels. The failure of this break-down is the result of glucose being converted to glycogen that is stored within the cells. Excess of this becomes toxic to the cell, thus affecting the organs in the body.
Diagnosis and Treatment
The diagnosis is often made at 4 -10 months of age. A blood test will usually reveal the low blood sugar level, accelerated levels of lipids and uric acid.
Treatment focuses on fixing the metabolic changes in the children and to assist them toward the progression of proper growth. Their diet may involve small amounts frequent feeds for the children though certain types of sugar may be restricted.




