Bioinformatics has expanded beyond just analyzing genome sequence data and is now used for various important tasks such as studying gene variation and expression, predicting and analyzing gene and protein structure and function, detecting gene regulation networks, creating simulation environments for whole-cell modeling, complex modeling of gene regulatory dynamics and networks, and presenting and analyzing molecular pathways to gain insight into gene-disease interactions.
Methods used for Creating Library (DNA to Data)
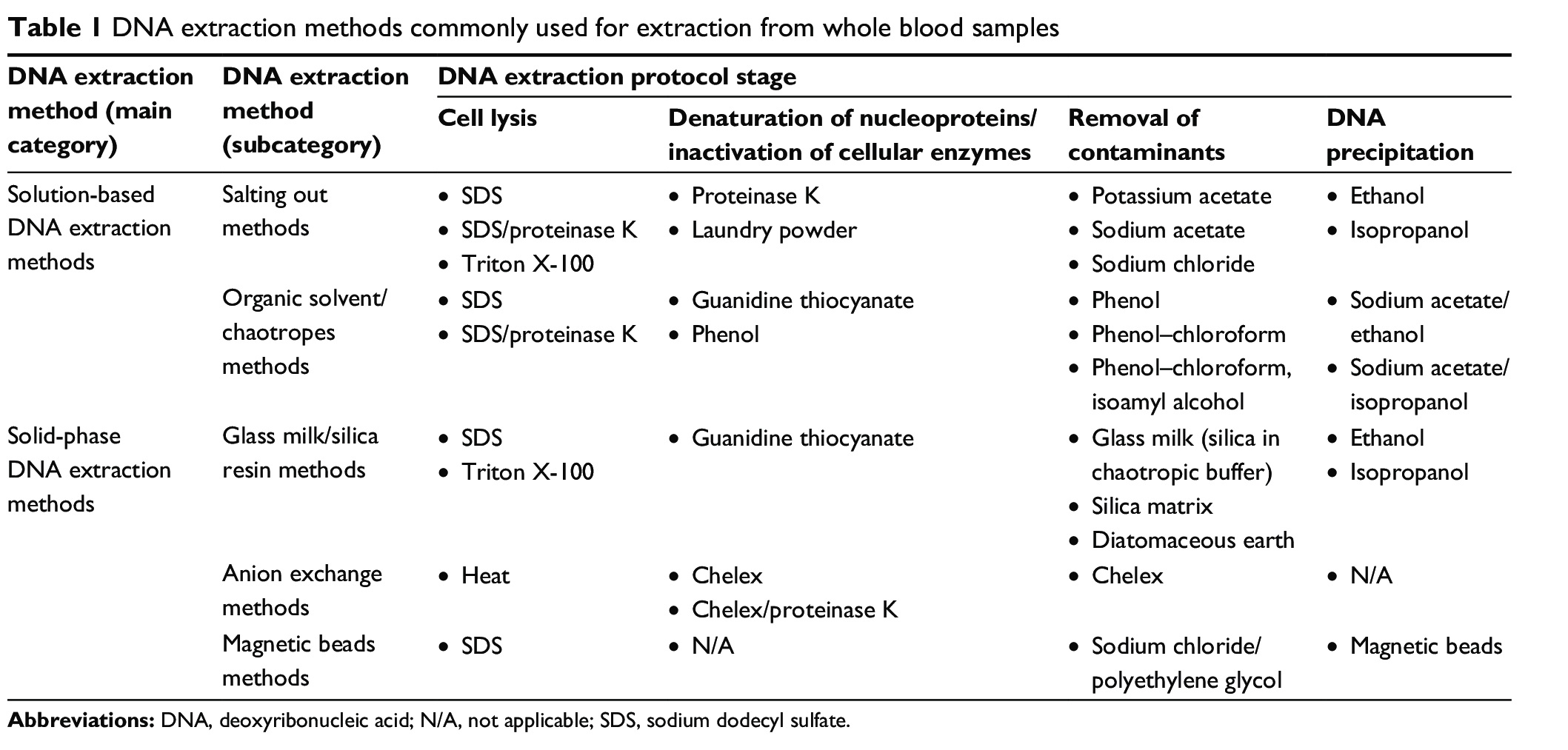
Sample Collection and DNA Extraction: Genomic DNA isolation is a fundamental step in many molecular biology experiments, including genome sequencing, gene expression analysis, and genetic engineering. The quality and quantity of the extracted DNA can significantly impact the downstream applications’ success, accuracy, and reliability. There are several methods available for DNA extraction, and the choice of method depends on the source of DNA, the downstream applications, and the required quality and quantity of DNA.
There are two primary solution-based approaches for DNA isolation:
1) Solution-based methods that use organic solvents, and
2) Those that rely on a solid base technique.
DNA Fragmentation: The genomic DNA is randomly fragmented into smaller pieces, typically ranging in size from 100 to 1000 base pairs. DNA can be shortened for library preparation using three common methods: Physical (using sound or pressure to break it into smaller pieces) -Sonication, enzymatic (using enzymes to cut or move the DNA), and chemical (breaking it down by heating it with certain metals).
Library Preparation: In the process of preparing DNA for sequencing, the broken ends of the DNA strands are first fixed to create flat, even ends. After that, tiny sequences called adapters are attached to the ends of the fragments. These adapters contain essential information needed for sequencing and enable the fragments to bind to the sequencing device.
The Nextera DNA Sample Prep Kit from Illumina is a method of preparing libraries of genomic DNA that involves using a transposase enzyme to fragment and label the DNA in a one-tube reaction known as “tagmentation.” This technique allows for the simultaneous fragmentation and tagging of the DNA, which simplifies and streamlines the library preparation process.
There are three strategies for optimizing assembly efficiency when preparing DNA libraries:
- Creating libraries with long inserts of approximately 1 kilobase in size. Avoiding PCR amplification during library preparation.
- Creating mate-pair libraries with a long distance spacing of 5-20 kilobases between reads.
- It is difficult to construct mate-pair libraries without PCR amplification, but long insert libraries can be constructed without PCR if there is enough DNA available. This is done by carefully breaking down genomic DNA into smaller pieces.
Sequencing: The prepared DNA library is loaded onto the sequencing instrument, and the DNA fragments are sequenced using one of several available sequencing technologies (e.g., Illumina, PacBio, Nanopore).
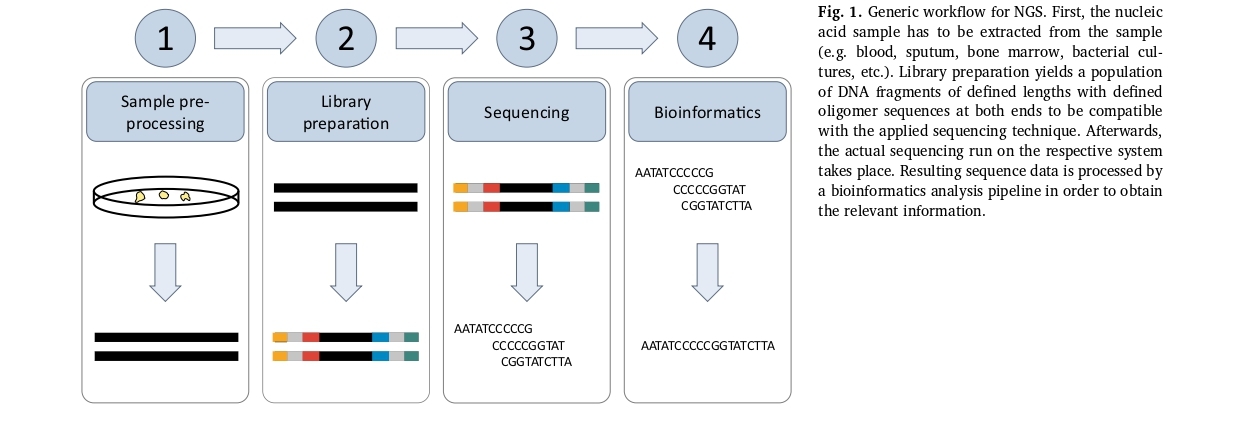
Next-generation sequencing (NGS) can be divided into four main process elements: sample preprocessing, library preparation, sequencing itself, and bioinformatics. Regardless of the sequencing method used, all modern sequencing technologies require a dedicated sample preparation step to create a sequencing library that can be loaded onto the instrument. The library consists of DNA fragments with a specific length distribution, which is tagged with oligomer adapters for barcoding, and then subjected to the actual sequencing process. Once the sequencing is complete, the resulting data is analyzed using bioinformatics tools.
During the library preparation, size selection and clean-up are essential steps to ensure that DNA fragments are of a specific length. Various methods can be used for this process, such as magnetic beads, columns, or gels. While solid-phase reversible immobilization beads are commonly used for size selection and clean-up, some companies offer spin-column clean-up kits, and the PACBIO SMRT bell Express Template Preparation Kit recommends gel-based size selection using the BluePippin System.
Polymerase chain reaction (PCR) is typically used to amplify nucleic acids during library preparation, both to attach sequencing adapters and to increase DNA concentration. However, amplification-free methods can lead to incomplete adapter attachment.
PCR amplification can result in bias, leading to under-representation or the complete absence of certain loci with extreme base compositions. Efforts have been made to reduce this bias, but special PCR workflows using unique molecular identifiers (UMIs) can be employed to avoid bias. These workflows involve a two to four-cycle UMI-PCR followed by an exchange of primers and a second PCR for amplification and adapter ligation. It is important to carefully automate these multi-step PCR workflows to prevent contamination during processing.
Data Processing: The raw sequencing data is processed to remove low-quality reads and adapter sequences, and to generate high-quality sequence data.
Base-calling: Base-calling is the procedure of determining the identity of nucleotides (A, C, G, T) from the signals produced by a sequencing device. These signals can be generated from various sources such as light intensity or electrical current fluctuations, which are associated with the passage of individual nucleotides through a nanopore. The base calling process involves matching these signals with specific nucleobases to generate a DNA sequence.
Quality control checks are performed to ensure sequencing data is sufficient followed by read mapping which is the process of aligning the sequencing reads and involves mapping them to a pre-existing reference genome or assembling them from scratch to create a preliminary assembly of the genome. It is accompanied by Variant calling, the process of detecting and describing the variations, including but not limited to SNPs, insertions, deletions, and structural variants, between the genome of the sample being sequenced and a known reference genome. After the variants have been identified, they are annotated to assess their possible effects on genes and other genomic elements. This process helps determine the potential functional consequences of the variants.
The identified variants are validated and analyzed using bioinformatics tools.
There are several bioinformatics tools used in genomic sequencing, and they vary depending on the specific analysis being performed.
Here are some commonly used tools:
Alignment and Assembly Tools: These tools are used to align sequencing reads to a reference genome or assemble reads to generate a de novo assembly. Examples include Bowtie2, BWA, and SPAdes.
Variant Calling Tools: These tools are used to identify and annotate variants between a sample and a reference genome. Examples include GATK, SAMtools, and FreeBayes.
Genome Annotation Tools: These tools are used to annotate genes and other genomic elements, such as regulatory regions and repeat sequences. Examples include ANNOVAR, Ensembl, and NCBI’s RefSeq.
Functional Analysis Tools: These tools are used to assess the functional impact of genetic variants on genes and other genomic features. Examples include SIFT, PolyPhen, and VEP.
Visualization Tools: These tools are used to visualize genomic data and aid in data interpretation. Examples include IGV, UCSC Genome Browser, and Circos.
Pathway and Network Analysis Tools: These tools are used to analyze the interactions between genes and proteins within a biological pathway or network. Examples include Reactome, KEGG, and STRING.
Machine Learning Tools: These tools are used to build predictive models using genomic data. Examples include Random Forests, Support Vector Machines, and Deep Learning algorithms.
Conclusion
In conclusion, the process of DNA to data in genome sequencing involves several steps, including sample preparation, DNA sequencing, data analysis, and interpretation. The advent of high-throughput sequencing technologies has revolutionized the field of genomics, enabling rapid and cost-effective sequencing of entire genomes. This has led to many exciting discoveries in various areas, including medicine, agriculture, and environmental science. However, there are still many challenges that need to be overcome, such as improving the accuracy and completeness of genome sequencing, reducing sequencing costs, and developing better tools for data analysis and interpretation. Despite these challenges, genome sequencing is a powerful tool that has the potential to transform many fields and improve our understanding of the genetic basis of life.
References
- Diego Chacon-Cortes, Lyn R Griffiths, “Methods for extracting genomic DNA from whole blood samples: current perspectives”, Journal of Biorepository Science for Applied Medicine, 2014
- Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, et al. Library construction for next-generation sequencing: overviews and challenges. BioTechniques. 2014;56: 61–64, 66, 68, passim. pmid:24502796, View ArticlePubMed/NCBIGoogle Scholar
- Adey A, Morrison Asan HG, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11:R119
- Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods. 2009;6:291–295. [PMC free article] [PubMed] [Google Scholar]
- Goodwin, S., McPherson, J. & McCombie, W. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333–351 (2016)
- Sage Science, 2019. BluePippin. Available from: [Internet]. http://www.sagescience. com/products/bluepippin/
- Richterich, Peter (1998-03-01). “Estimation of Errors in “Raw” DNA Sequences: A Validation Study”. Genome Research. Cold Spring Harbor Laboratory. 8 (3): 251–259. doi:10.1101/gr.8.3.251. ISSN 1088-9051. PMC 310698. PMID 9521928.




