The genome is a complex set of genetic information present in the nucleus and mitochondria of eukaryotic cells, including humans. Mitochondrial DNA was discovered and sequenced in 1981, while the draft sequence of the human genome published in February 2001. The highest quality representation of the human genome is the GRCh38/hg38, released in December 2013 by the Genome Reference Consortium (GRC). This assembly version is superior because it addresses issues such as gaps, variants, component, and tiling path errors, and includes sequence-based representations of centromeres and telomeres for the first time. The Human Genome Project (HGP) was completed in 2003, costing around $2.7 billion. The project relied on automated Sanger sequencing and hierarchical assembly of large insert clones to sequence the genome. The phrase “the human genome” often emphasizes the high level of similarity between individuals but it tends to overlook the millions of genetic differences that make each of us unique. However, the field of human genetics is interested in understanding how our genetic variations contribute to our physical and biological differences, rather than focusing solely on our similarities.
The genome contains all the genetic information of an organism or cell, stored in linear or circular sequences of nucleic acids. To determine this sequence, technologies have been developed that are more accurate, faster, and have higher throughput. However, the sequencers generate sequences that are usually shorter than the genome being investigated, so the complete genome sequence must be deduced by assembling these shorter fragments. In the past, only one individual per species was sequenced due to time and cost constraints, and this individual’s sequence became the reference genome for the species. Reference genomes are crucial for downstream applications such as gene manipulation experiments and studying molecular evolution. Next Generation Sequencing (NGS) platforms were developed in the early 2000s to address larger genomes in a process called Whole Genome Sequencing (WGS), which has become increasingly more efficient and affordable. New sequencing technologies have emerged that promise to generate higher-quality genomes.
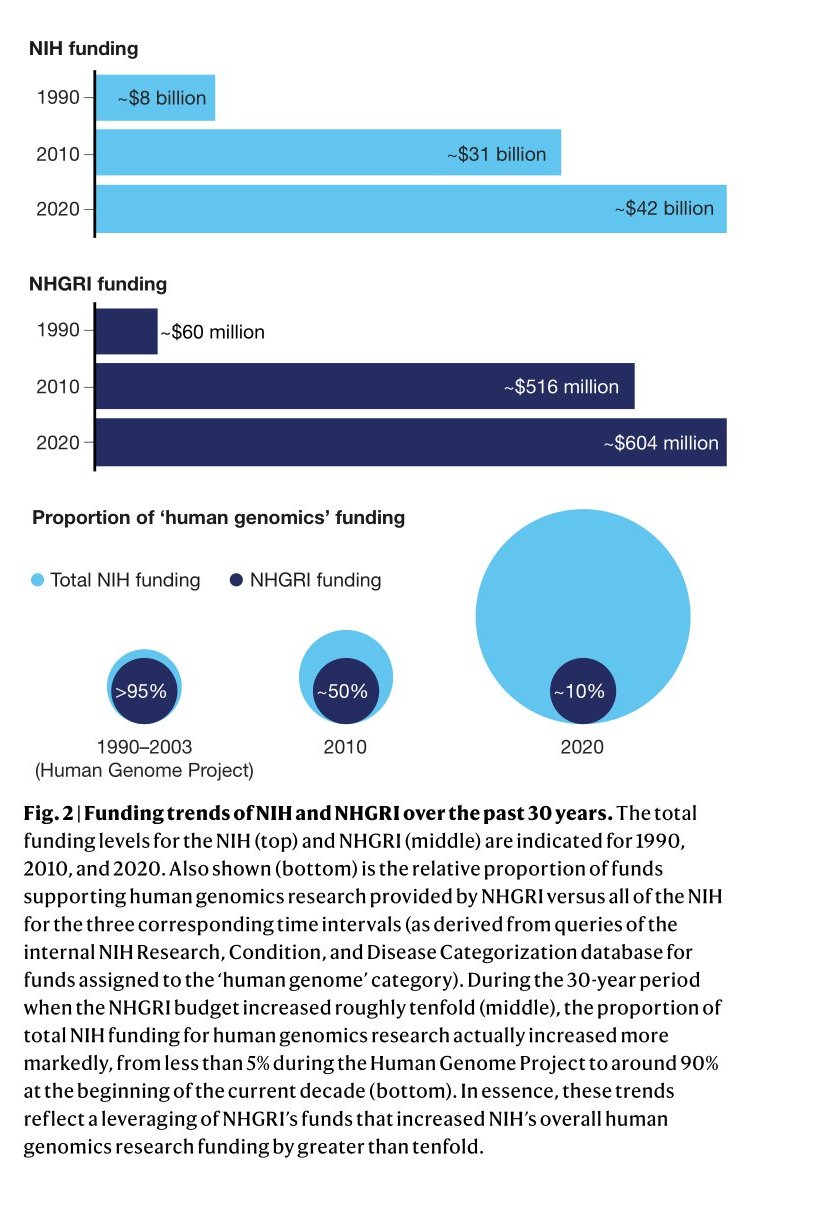
The frequent use of the word “the” with “human genome” highlights how closely similar individual humans are to each other (99.9%), but it ignores the fact that each person has millions of genetic differences (0.1%) that make them unique. However, the reason for the existence of human genetics as a field is not to focus on our similarities but to understand how our genetic variations contribute to our physical and behavioral differences. Genomic sequencing is a laboratory technique used to analyze the complete genetic material of a particular organism or cell type. It is useful in identifying alterations in specific regions of the genome, which can provide insight into the development of diseases like cancer.
Grand Challenges in Genomics Research
There are various potential grand challenges that the field may face in the future, such as comprehending the function of each gene in the human genome. Scaling up the identification of causal variants, genes, and mechanisms for existing GWAS signals from a few to thousands is also one of these challenges. In addition, creating a comprehensive molecular atlas of all human cell types from birth to death, which includes their chromatin landscape, gene expression, and protein expression signatures while being spatially resolved, is another significant challenge. Another crucial task is developing precise and quantitative models that can predict the impact of arbitrary sequence variants on gene expression and/or protein function in any of these cell types.
Advancements in Genomics Research
Other significant challenges in the field include the development of effective treatments for genetic diseases that are currently untreatable. Additionally, achieving a more comprehensive understanding of the non-coding regions of the genome and their involvement in gene regulation is essential. Ethical guidelines should also be established for the appropriate use of genomic data and technologies in both research and clinical practice. Furthermore, developing innovative technologies for genome sequencing, analysis, and manipulation is crucial. It is also important to build global collaborations and resources to facilitate the sharing of genomic data and knowledge. Lastly, integrating genomic data with other omics data such as transcriptomics, proteomics, and metabolomics is necessary to gain a more complete understanding of biological systems.
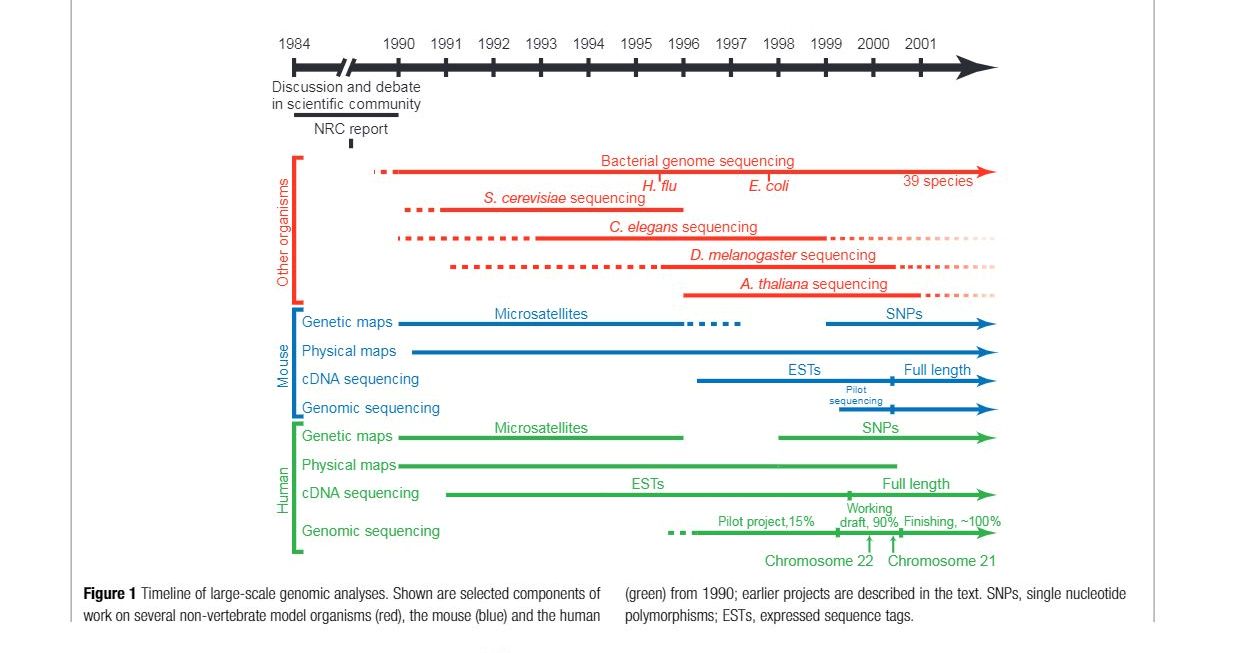
Timeline of Large Scale Genome Analysis
The first significant achievements in genome research were the creation of genetic and physical maps of the human and mouse genomes, which helped identify disease genes and served as reference points for genomic sequencing. The second breakthroughs were the sequencing of the yeast and worm genomes, as well as specific regions of mammalian genomes. These projects demonstrated the feasibility of large-scale sequencing and established a two-phase approach for genome sequencing. In the first phase, called “shotgun,” the genome is divided into manageable segments, and each segment is thoroughly covered through the sequencing of randomly selected subfragments. In the second “finishing” phase, any gaps in the sequence are filled, and any remaining uncertainties are resolved through targeted analysis. These efforts revealed that complete genomic sequencing provides valuable information about genes, regulatory regions, and chromosome structure that cannot be obtained through cDNA studies.
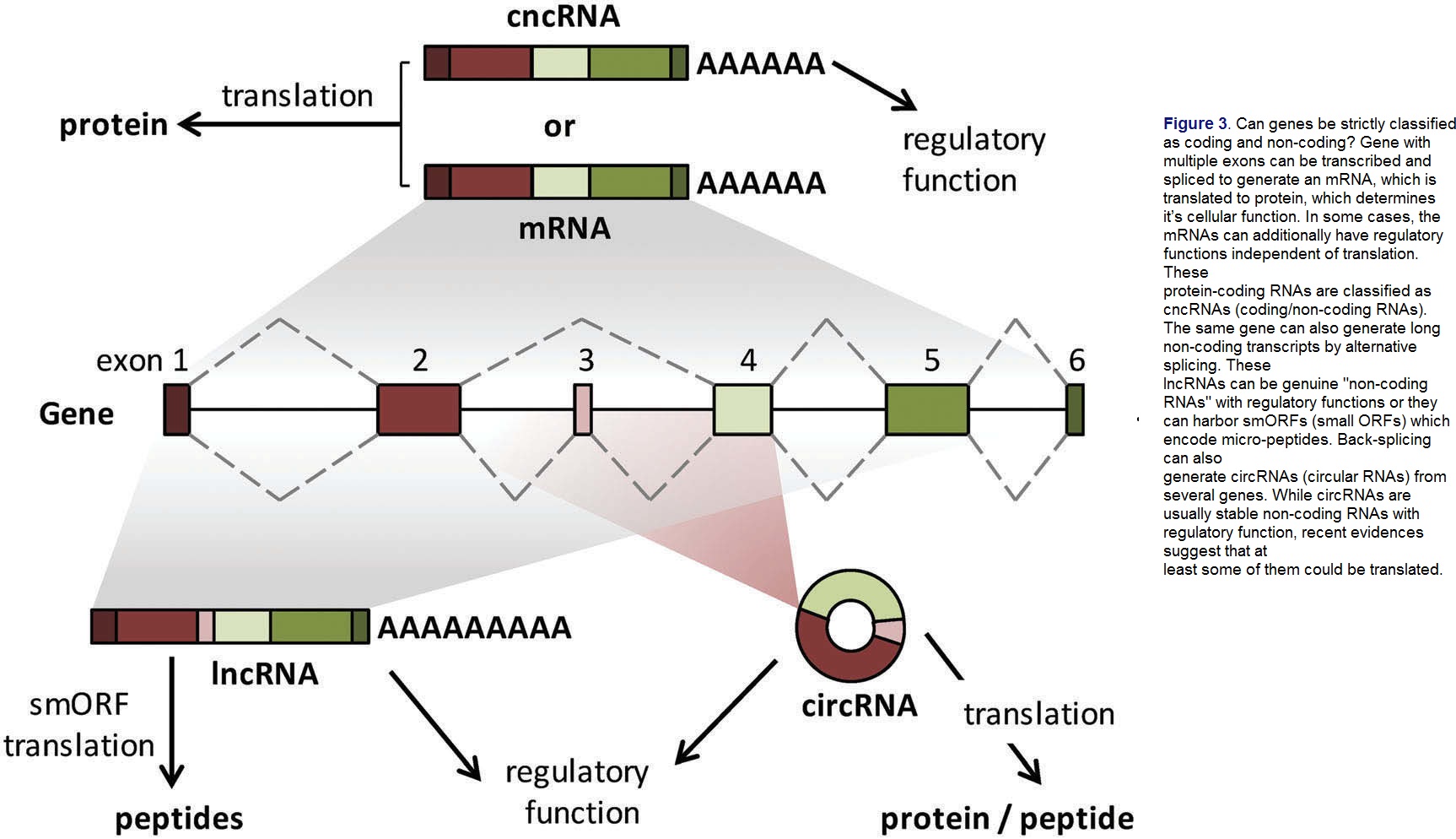
Earlier expectations were that the human genome would have a larger number of protein-coding genes due to higher eukaryotic complexity. However, it was surprising to discover that the human genome only contains around 30,000 protein-coding genes, similar to the worm genome. This was due to undervaluing the non-coding parts of the genome, which make up over 95% of the human genome. Short and long non-coding RNA genes, including lncRNAs, play critical roles in unique processes such as neuronal development and cancer metastasis. Alternative splicing contributes to functional diversity and complexity in both the proteome and non-coding transcriptome. Splicing and posttranscriptional processing are crucial in mammalian physiology and pathology.
The traditional classification of genes relied on the presence of long, conserved, and translatable open reading frames (ORFs). However, recent discoveries have revealed that some non-coding RNAs encode for short micro peptides. Gene classification can also be based on their biological function, but the discovery of cncRNAs, which have both coding and non-coding functions, challenges this approach. For instance, the mRNA encoding the tumor suppressor protein p53 has a non-coding function that directly inhibits MDM2 activity. This blurs the distinction between coding and non-coding genes.
General Themes for the Regulatory Role of the Noncoding Transcript Variants
Changes in noncoding DNA regions can also cause health problems by altering gene activity or protein assembly. Variants in noncoding DNA can turn on or off genes, leading to disrupted development or diseases such as cancer and developmental disorders. Noncoding DNA includes enhancers, promoters, insulators, silencers, and functional RNA molecules. Genetic alterations in noncoding DNA can be inherited or acquired during a person’s life. The potential of non-coding transcript variants of protein-coding genes to act as genuine lncRNAs with scaffolding functions. It presents examples of genes such as SRA1 and ASCC3 and explains the different functions that can be attributed to non-coding transcripts arising from hybrid genes, including regulation of splicing and mRNA processing, miRNA sponge activity, competitive RBP-binding/sequestration, and micro peptide/microprotein expression. The passage also discusses the role of intron retention and back-splicing of circRNAs in modulating gene expression, and the ability of lncRNAs and circRNAs to function as ceRNAs or miRNA sponges. Finally, the passage presents the idea that a large proportion of lncRNA products from bifunctional or hybrid genes could express micro peptides that are functionally related to their canonical mRNA-encoded proteins.
The discovery of human-specific de novo genes has had weak links to disease in the past, but recent research has found a de novo gene called NYCM, present exclusively in humans and chimpanzees, to be involved in the development of neuroblastoma. Another study found that a transcript containing a de novo open reading frame (ORF) that originated from an endogenous retrovirus is essential for maintaining pluripotency. These findings are the first experimental evidence of the functional importance of de novo genes in humans.
In recent years, there has been growing interest in RNA molecules due to the discovery of many noncoding genes, which has expanded our understanding of their functions in organisms. One interesting finding is the presence of peptides encoded by long noncoding RNAs, which have revealed new mechanisms for these molecules. Although thousands of these peptides have been identified in human cells, their roles are still largely unknown. Recent discoveries of micro peptides and their involvement in various biological processes, as well as the potential regulatory functions of noncoding mRNAs are emerging.
Non-coding genes, also known as non-coding RNAs, have various functions in the regulation of gene expression. They can act as transcriptional regulators, helping to control when and where genes are expressed. They can also act as post-transcriptional regulators, controlling the processing and stability of messenger RNAs (mRNAs) before they are translated into proteins.
Some non-coding RNAs, such as microRNAs, can bind to specific mRNAs and prevent them from being translated into proteins. Others, like long non-coding RNAs, can act as scaffolds or guides, bringing together different components of the gene expression machinery and helping to regulate protein production.
In addition to their roles in gene regulation, non-coding RNAs have also been implicated in a wide range of biological processes, including development, cell differentiation, and disease. They have been linked to various types of cancer, neurological disorders, and immune system dysfunction, and are an active area of research for understanding the molecular basis of these diseases.
Advancements in Human Genome Assembly
T2T-CHM13 refers to a high-quality human genome assembly generated using long-read sequencing technology. CHM13 is the name of the human haploid cell line used to generate the assembly. The T2T-CHM13 assembly has added five full chromosome arms and more additional sequences than any previous human genome reference release in the past 20 years. Technological limitations in the past have prevented the comprehensive study of genomic variation across the entire human genome, but high-accuracy long-read sequencing has overcome this barrier. However, a different sample type will be required to complete the last remaining chromosome, the Y chromosome. Long-read resequencing studies are needed to comprehensively survey polymorphic variation in the most repetitive regions of the genome. To address the bias of CHM13 not capturing the full diversity of human genetic variation, the T2T Consortium and the Human Pangenome Reference Consortium are collaborating to build a collection of high-quality reference haplotypes from diverse samples. Although automated T2T assembly of diploid genomes presents a challenge, the T2T-CHM13 assembly represents a more complete, representative, and accurate reference than GRCh38. GRCh38 is a reference genome assembly for the human genome, released by the Genome Reference Consortium in December 2013. It is an updated version of the GRCh37 assembly (also known as hg19), which was released in 2009.
GRCh38 includes improvements in sequencing and assembly technologies, as well as updates to the annotations of genes and other genomic features. It is a more complete and accurate representation of the human genome than previous assemblies.
GRCh38 is widely used as a reference for genomic research and is the most current version of the human genome assembly. It contains over 3 billion base pairs and is organized into 24 distinct chromosomes, with additional unplaced contigs representing regions that could not be reliably assigned to a specific chromosome.
Genomics and Agriculture
Laboratory discoveries in genomics have rapidly made their way into commercial agriculture, with companies utilizing these advances. It is anticipated that in the future, genomic selection based on thousands of gene markers will be used for a wider range of economically important traits instead of traditional selection methods based on estimated breeding values (EBVs). As further discoveries and understanding of livestock genomes are made, it will only increase their potential to provide sustainable protein worldwide and contribute to biomedical research. To fully realize the impact of genomics on animal agriculture, smart and environmentally sustainable policies, access to technology for all levels of producers, and continued financial support for short- and long-term genomics research will be necessary.
Predictions for the Future of Human Genomics by 2030
Here are some predictions for the future of human genomics that may come true by 2030. Although not all of these predictions may be fully achieved, striving toward them could lead to significant advancements.
- Generating and analyzing a complete human genome sequence will become routine for any research laboratory, like a basic DNA purification process.
- It is necessary to have an understanding of the biological function of every human gene and to consider knowledge about non-coding elements in the genome as a standard.
- The epigenetic landscape and transcriptional output will be routinely incorporated into predictive models of how genotype affects phenotype.
- Human genomics research will move beyond population descriptors based on race.
- Studies involving genome sequencing and phenotype information for millions of participants will be commonplace in school science fairs.
- Genomic testing will be a routine procedure in clinical settings, similar to complete blood counts.
- The clinical relevance of all genomic variants will be easily predictable, making the term “variant of uncertain significance” obsolete.
- An individual’s complete genome sequence and annotations will be accessible on their smartphone if desired.
- Advancements in human genomics will benefit individuals from diverse backgrounds equitably. Breakthrough discoveries will lead to curative therapies for genetic diseases involving genomic modifications.
References
- Shendure, J., Findlay, G. M., & Snyder, M. W. (2019). Genomic Medicine–Progress, Pitfalls, and Promise. Cell, 177(1), 45–57. doi:10.1016/j.cell.2019.02.003
- Giani, A. M., Gallo, G. R., Gianfranceschi, L., & Formenti, G. (2019). Long walk to genomics: History and current approaches to genome sequencing and assembly. Computational and Structural Biotechnology Journal. doi:10.1016/j.csbj.2019.11.002 10.1016/j.csbj.2019.11.002.
- Eric D. Green, et.al, “Strategic vision for improving human health at The Forefront of Genomics”
- Sonam Dhamija, ”Non-coding transcript variants of protein-coding genes – what are they good for?”, RNA BIOLOGY,2018, VOL. 15, NO. 8, 1025–1031, https://doi.org/10.1080/15476286.2018.1511675
- McLysaght A, Guerzoni, ” New genes from the non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation. Phil. Trans. R. Soc. B 370: 20140332
- Alexander et.al; ,”Non codingRNA:whatisfunctionalandwhatisjunk?:, Department ofBiochemistry,UniversityofToronto,Toronto,ON,Canada
- Sergey Nurk et al. “The complete sequence of a human genome”, Science376, 44-53(2022).DOI:10.1126/science.abj6987.




