In the study of bacterial communities using DNA analysis, a technique called culture-independent analysis is commonly used. This method involves using next-generation sequencing machines to analyze the DNA present in a sample. This DNA can come from living bacteria, dead bacteria, or even free-floating DNA fragments. The presence of DNA from deceased bacteria may cause difficulties in accurately detecting the types of living bacteria in a sample, thus affecting the analysis process.
Recent technological advancements in science have allowed us to increase our comprehension of the diversity and prevalence of minute organisms that form the microbiota, along with their interactions with the environments they inhabit. Nucleic acid sequencing technologies have provided access to previously undetected microorganisms that could not be grown in laboratory culture. With the availability of rapid and inexpensive sequencing platforms, it is now possible to sequence the genomes of dozens of strains of a single microbial species or to analyze vast communities of microbiota from various environments.
These advancements, along with concurrent investments in microbial ecology, evolution, forensics, and epidemiology, have transformed our ability to use genomic sequence information to investigate the origins, evolution, and catalysts of historical, emergent, and reemerging disease outbreaks. Nucleic acid sequencing has proven to be a valuable tool in improving disease surveillance, detection, and response efforts by offering valuable insights into the extensive diversity and intricacy of microbial life and their connections with the environment.
Recently, whole genome sequencing has been used to investigate outbreaks of emerging, reemerging, and novel infectious diseases. Through the comparison of genomic sequences of strains that are closely related, researchers have been able to trace the evolution of isolates during a disease outbreak, track the transmission of a communicable disease from person to person, and pinpoint the sources of disease outbreaks. This knowledge has also helped in the identification of factors that contribute to the emergence, virulence, or spread of pathogens and in accelerating the development of diagnostic tools. An example of this is the recent use of rapid genome sequencing to halt the spread of a methicillin-resistant Staphylococcus aureus (MRSA) infection in a neonatal ward at a hospital located in Cambridge, UK.
Genome Sequencing and the Importance of Studying Microbial Communities
The term “microbiome” refers to both the microbial community and the environment it inhabits, similar to how “biome” is used in ecology. “Microbiome” can also refer to the collective genomes of microbes, which was originally coined by Joshua Lederberg. However, “microbiome” is commonly used to describe the collection of microbiomes in the human body, with the gut microbiome being a specific example.
Previously, research on microbes in human health focused on pathogenic organisms, with limited attention given to communities of non-pathogenic microbes. However, microbiome research has highlighted the importance of understanding the genomes and behavior of these microbes, as they provide important functions such as digestion and protection against infection. Antibiotic treatment can disturb the microbiome, leading to infections and the overgrowth of antibiotic-resistant organisms. It is essential to understand the structure and behavior of microbial communities to manipulate the microbiome and treat diseases effectively. Many organisms have not been cultured and depend on other community members for growth requirements, making the study of individual organisms reliant on studying the community as a whole.
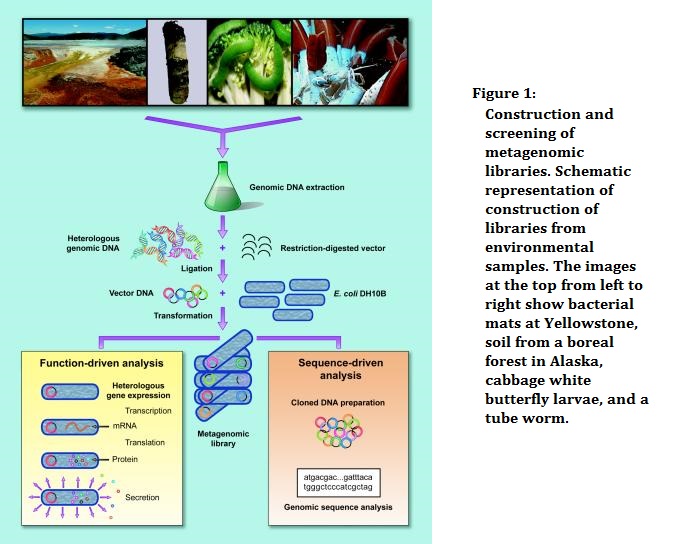
Dissecting a Microbiome
To analyze the structure of microbial communities, researchers can either focus on specific regions like the 16S rRNA gene or conduct shotgun sequencing to identify present genes. They can also sequence genomes of individual organisms to create a reference genome catalog and analyze RNA to identify RNA viruses and describe the transcriptome.
Methods of Sequencing Technologies used in Microbial Communities
Next-generation sequencing (NGS) has revolutionized the analysis of DNA sequences, allowing for high-throughput analysis of PCR amplicons or environmental nucleic acids, and has led to the development of new water quality assessment methods. In clinical research, NGS has been used as a screening tool for detecting and identifying disease-causing agents in place of conventional diagnostic methods such as culturing or microscopy. While quantitative tests like qPCR and culture-based FIB quantification kits are suitable for estimating exposure to biological risk agents or sewage contamination, NGS surveys can be used as a first step for more specific exposure assessment of appropriate targets.
Considerations in Choosing a Next-Generation Sequencing (NGS) Method for Microbiome Analysis
Some common considerations in choosing an NGS method for microbiome analysis include taxonomic resolution, functional profiling, host contamination, false positives, bias, and post-sequencing computational requirements.
16S rRNA sequencing, shotgun metagenomic sequencing, and RNA sequencing can all be used to determine what bacteria are present in a microbiome, but shotgun metagenomic sequencing and RNA sequencing can also detect members of other domains, such as fungi, parasites, and viruses. Only RNA sequencing can examine RNA viruses.
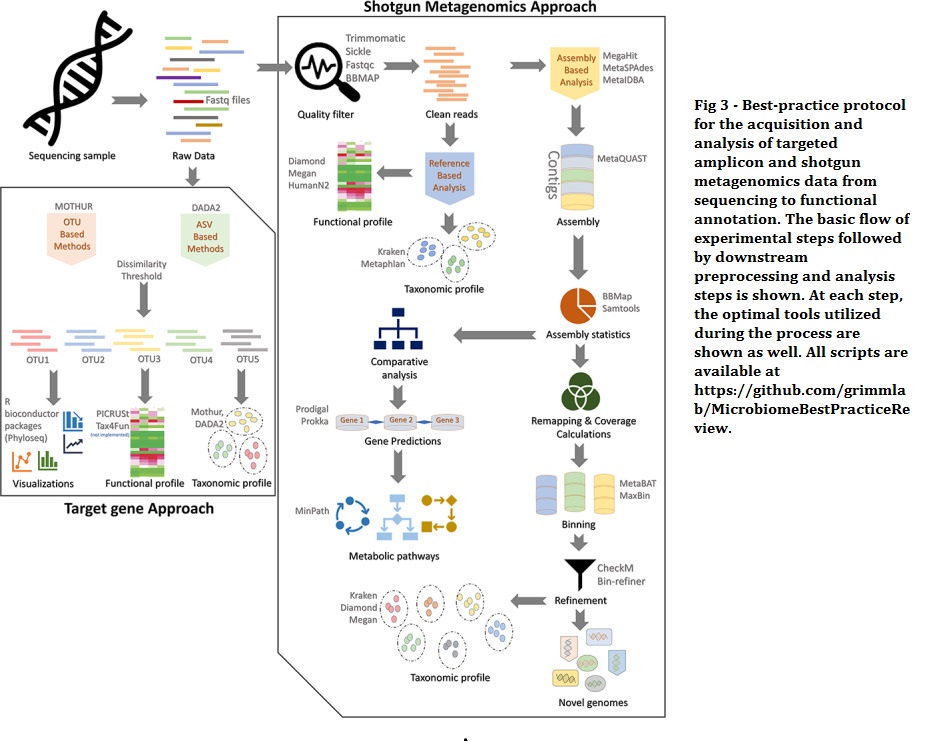
An overarching finding of studies that have compared these methods is that phylum designations are comparable, but 16S rRNA sequencing tends to offer less resolution and sensitivity for detecting changes at the species level and cannot detect strain-level changes.
Shotgun metagenomic sequencing generally results in the improved genus- and species-level classification. Functional profiling cannot be directly obtained from 16S rRNA sequencing, but methods like PICRUSt or Tax4Fun aim to predict functional profiles of bacteria based on 16S rRNA data. However, the success of these methods, when compared with functional potentials obtained via shotgun metagenomics, varies with the 16S gene primers used for amplification. Conversely, shotgun metagenomics and RNA sequencing consider all the microbial DNA and RNA, thus allowing for a more comprehensive prediction of functional potential. RNA sequencing identifies which genes are actively being transcribed (active functional profile), while shotgun metagenomics provides a random selection of all genes encoded by the microbes (predictive functional potential).
There is less risk of host contamination and false positives in 16S rRNA sequencing compared with other NGS methods because the gene being amplified and sequenced (i.e., the 16S rRNA gene) is specific to bacteria. However, there is a higher risk of bias with 16S rRNA sequencing due to primer-dependent PCR amplification bias and differences between the variable regions.
Finally, the cost must be considered for any project, and the differences in cost between the methods relate to the amount and depth of sequencing. Shotgun metagenomics and RNA sequencing analyses typically require much more sequence data than 16S rRNA sequencing, resulting in higher costs. However, 16S rRNA sequencing is more accessible to researchers with beginner- and intermediate-level bioinformatics experience.
Taxonomic Profiling
Different methods for identifying species from a dataset of complete prokaryotic genomes are available that includes SpeciesFinder was used as a baseline as it relied solely on the 16S rRNA gene, the second method, Reads2Type, searched for species-specific 50-mers, primarily within the 16S rRNA gene, using non-species-specific 50-mers to quickly narrow down the search, rMLST, predicted species by analyzing 53 ribosomal genes, TaxonomyFinder, relied on species-specific functional protein domain profiles. Finally, KmerFinder predicted species by analyzing the number of overlapping 16-mers.
Functional Profiling
The functional composition of a microbial community’s metagenome includes the process of two steps: the first step, called “gene content inference,” precomputes the gene content for each organism in a reference phylogenetic tree. This creates a table of predicted gene family abundances for each organism in the 16S-based phylogeny. This step is performed once and is independent of any particular microbial community sample.
The second step, called “metagenome inference,” combines the resulting gene content predictions for all microbial taxa with the relative abundance of 16S rRNA genes in one or more microbial community samples. This step corrects for the expected 16S rRNA gene copy number and generates the expected abundance of gene families in the entire community. In other words, this approach uses the 16S profile to infer the functional potential of the microbial community, allowing for insights into the metabolic capabilities and potential interactions between different microorganisms in the community.
Comparing and Clustering Microbial Genomes
Operons are important for controlling gene expression in bacteria and several algorithms are available to predict them. However, very few algorithms efficiently study gene clusters across hundreds of genomes. Lee and Sonnhammer (2003) proposed a querying strategy to analyze gene clusters across a large number of genomes. They analyzed gene clustering in 400 bacterial genomes by starting from a well-characterized list of operons in Escherichia coli K12. They validated their algorithm by comparing the results to experimentally verified operons in Bacillus subtilis subsp. subtilis str. 168 genome and E. coli K12 genome. They performed a comparative analysis of operon occurrences among bacterial groups, studied gene orientations within predicted clusters, and analyzed distributions of rearrangements both within and across clusters. Their algorithm is well suited for analyzing gene clusters across a large number of genomes and provides important biological insights.
Challenges
New sequencing technologies have led to an increase in computational tools for analyzing biological data, but challenges still exist due to data complexity, lack of metadata, and standard data formats. It is important to benchmark, and make tools open-source and easy to install with a proper user interface for reproducibility and interpretation of results.
While recent computational developments offer scalable solutions, it’s still important to implement multiple high-throughput strategies to ensure the accuracy of genomic findings.
To achieve a more precise description of genomes and their environmental functions, sampling saturation biases must be addressed by improving the resolution of genomic analysis. To accomplish this, more in-depth analyses of low-complexity communities using metatranscriptomics and metaproteomics technologies are necessary.
Metatranscriptomics involves analyzing community transcripts directly from different environments to correlate taxonomic signatures with functions by profiling mRNA transcripts generated under various environmental conditions. Combining shotgun metagenomics with metatranscriptomics can aid in achieving higher-resolution analysis. Metaproteomics, on the other hand, involves analyzing microbiome-associated protein profiles to provide information on function under different environmental conditions. However, community protein profiling relies heavily on the accuracy of metagenomics data.
Mass spectrometric analysis of peptides generated from an environmental sample can be matched with predicted proteins from metagenomics analysis. In conclusion, the future of target gene and metagenomics projects not only depends on emerging computational resources but also more in-depth and complementary sequencing methodologies to establish more comprehensive approaches for delineating the functional profiles of environmental samples.
References
- Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004 Dec;68(4):669-85. doi: 10.1128/MMBR.68.4.669-685.2004. PMID: 15590779; PMCID: PMC539003.
- Hosokawa M, Endoh T, Kamata K, Arikawa K, Nishikawa Y, Kogawa M, Saeki T, Yoda T, Takeyama H. Strain-level profiling of viable microbial community by selective single-cell genome sequencing. Sci Rep. 2022 Mar 15;12(1):4443. doi 10.1038/s41598-022-08401-y. PMID: 35292746; PMCID: PMC8924182.
- Kumar, N. V., Menon, T., Pathipati, P., and Cherian, K. M. (2013). 16S rRNA sequencing as a diagnostic tool in the identification of culture-negative endocarditis in surgically treated patients. J. Heart Valve Dis. 22, 846–849.
- Wensel CR, Pluznick JL, Salzberg SL, Sears CL. Next-generation sequencing: insights to advance clinical investigations of the microbiome. J Clin Invest. 2022 Apr 1;132(7):e154944. doi: 10.1172/JCI154944. PMID: 35362479; PMCID: PMC8970668.
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW, 2007, RNAmmer: “Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100 –3108. http://dx.doi.org/10.1093 nar/gkm160.
- Langille, M., Zaneveld, J., Caporaso, J. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821 (2013). https://doi.org/10.1038/nbt.2676.
- Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2021 Jan 18;22(1):178-193. doi: 10.1093/bib/bbz155. PMID: 31848574; PMCID: PMC7820839.