Monoclonal Antibody
Monoclonal antibodies (mAbs) are antigen-recognizing glycoproteins. They are made of identical immune cells and all are clones of a unique parent cell. Since 2014, FDA has approved at least five monoclonal antibodies per year, and this trend shows no signs of slowing. Monoclonal antibody therapy encompass a number of clinical conditions such as autoimmune disorders, infectious diseases, and oncology, among others.
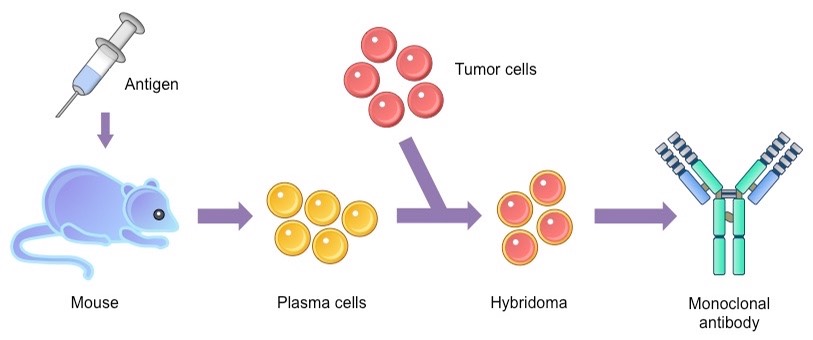
Hybridoma technology is a method used for producing large number of identical antibodies, also referred as monoclonal antibodies. Immunological response is provoked in a mouse by injecting an antigen. The B cells (type of white blood cell) that produces antibodies (which binds to the antigen) are harvested and fused with identical cancer cells, referred as myeloma cells. The fused cells, hybrid cell line, hybridoma, has both the antibody-producing ability of the B-cell and longevity and reproducibility of the myeloma.
This hybrid cell line, or hybridomas is grown in culture and each culture consists of genetically identical hybridomas producing the same antibody thus referred as monoclonal antibodies. Researchers who need this workflow carried out for a specific, uncatalogued target can outsource the entire process through a custom monoclonal antibody development service rather than building hybridoma capability in-house. When cultures produce more than one antibody or different antibodies, it is referred as polyclonal antibodies. The myeloma cell line is carefully selected for its ability to grow in tissue culture and for an absence of antibody synthesis. Below shown is a pictorial representation of the same.
History
In the year 1798 Edward Jenner demonstrated first smallpox vaccination, his apparent discovery was met with much opposition and even ridiculed as he could not explain how and why the vaccine worked. In time the value of his vaccine was recognized, but as many poorer communities had limited access to medical treatment it was several decades before its full benefits were realized. It was only in 1853, 30 years after Jenner’s death, smallpox vaccination was made compulsory in England and Wales.
Nearly a century later, Emil Roux’s discovery of diphtheria toxin influenced Emil Von Behrin a German physician and one of the founders of Immunology. In 1890, he showed that it was possible to provide an animal with passive immunity against tetanus by injecting it with the blood serum of another animal infected with the disease. This lead to the development of first antitoxin for diphtheria and Emil Von Behrin antitoxin was the first effective treatment for this deadly disease. In 1901,Emil Von Behring received the Nobel Prize for the diphtheria antitoxin he developed. As von Behring’s serum therapy was only useful against a small number of diseases. Despite his important role in reducing diphtheria, which his work was eventually overshadowed by the work of Paul Ehrlich side chain theory leading to contributions in immunology that resonate today. Later in his career, he applied the same theory to the study of tumor cells and effectively invented the field of chemotherapy, a word which he coined.
The term Antikörper (‘antibodies’) was introduced by Paul Ehrlich’s. In his experimental studies on Immunity in 1891, he fed the mice with toxin and started testing their resistance to different amounts of that particular toxin. Ehrlich gradually increased the doses for individual mice until they could resist doses that would have been lethal if they had been administered straight away. Even more impressively, Ehrlich eventually produced mice that could survive doses of ricin, (one of the world’s deadliest toxin )that were hundreds of times stronger than the lethal dose for mice that hadn’t been exposed, making it clear that something in the immune system of the mice was developing resistance to the toxin.
Based on his experiments and findings Ehrlich put forth his ‘magic bullet’ theory, also known as the ‘side-chain’ theory. The underlying concept was similar to smallpox vaccination, introduced by Edward Jenner in 1798. What separated Ehrlich’s work from Jenner’s was his insights into the mechanisms of immunity, particularly the side-chain theory that he proposed in 1900. The theory suggested that side chains within the cells react with antigens and bind them – fitting like a lock and key – to create antibodies, which then travel around the body in the blood.
After the introduction of Ehrlich’s side-chain theory, researchers continued to work on antibodies. In 1948, Astrid Fagraeus described the role of plasma B cells in the production of antibodies. Later in 1959, Gerald Edelman and Rodney Robert Porter published their independent discoveries about the chemical structure of antibodies, which led to them jointly receiving the 1972 Nobel Prize in Physiology or Medicine. Both Edelman’s work and Porter’s focused on breaking antibodies down into smaller parts so they could be handled and studied more easily, which had important implications for diagnostics and therapy. Porter used the enzyme papain to split antibody molecules into three parts, and Edelman focused on breaking the disulphide bridges holding the molecule together. Their work revealed that antibodies consist of two pairs of chains – two short ‘light’ chains and two ‘heavy’ chains, which are about twice the length of the light chains – held together in a ‘Y’ shape by disulphide bridges. The work paved the way for the development of monoclonal antibody therapy, which is used today to treat cancer and autoimmune diseases.
In 1973, the first atomic resolution structure of antibody was published and the scientific community were now aware of the vast diverse types of antibodies produced by the immune system. One of the main issue that was restricting further research in antibody at the time was the inability to isolate and purify single antibodies of known specificity from the billions produced in the body. The answer to this was given by Georges Kohler and Cesar Milstein who invented a technique of forcing immune system cells to make pure antibodies against a chosen antigen. The invention was made in 1974 when Dr. Kohler, then a young postdoctoral student, was working with his mentor, Dr. Cesar Milstein at the Laboratory of Molecular Biology in Cambridge, England.
Kohler and Milstein had created a powerful general method for raising pure antibodies also known as monoclonal antibodies against any antigen of interest. In principle these pure antibodies known as, because of their remarkable specificity, could be used to diagnose disease, or to carry therapeutic agents to particular body tissues.
Köhler and Milstein developed the hybridoma method of murine antibody production in 1975, which allowed the production of the first mAb to market; Johnson & Johnson’s Orthoclone OKT3 (muromonab) in 1986. The discovery of monoclonal antibodies (mAbs) produced by “hybridoma technology” by George Köhler and Cesar Milstein in 1975 has had a great impact both on basic biological research and on clinical medicine. However, this impact was not immediately recognized. The British Government, which supported the laboratory for Köhler and Milstein study, did not recognize the extraordinary commercial significance of the technique and failed to patent it. It took around 10 years to appreciate the importance of using these mAbs in various fields of science other than immunology, such as cell biology, biochemistry, microbiology, virology, para-sitology, physiology, genetics, and molecular biology; and also in areas of clinical medicine, such as pathology, hematology, oncology, and infectious disease. The contribution of mAbs to science and clinical medicine was recognized in 1984 by the award of the Nobel Prize for Medicine to Köhler and Milstein.
The discovery of murine mAbs by Kohler and Milstein paved the way for further developments in antibody engineering technology during 1990 and was followed by regulatory approvals for several therapeutic mAbs like Rituximab in 1997, trastuzumab 1998, alemtuzuman in 2001, bevcizumab in 2004, cetuximab in 2005, ofatumumab in 2009, perfuzumab ini 2012, ramuciruman in 2014 and secukinumb in 2015.
Although there were high expectations for murine mAbs, due to problems with immunogenicity they have declined from favor. Murine antibodies are seen as foreign by the immune system, which mounts a response known as the Human Anti-Mouse Antibodies (HAMA) response, producing unwanted side effects. In an attempt to overcome the HAMA response antibodies were developed that were chimeric; consisting of 60% human and 40% mouse immunoglobulin. The next step in the development of mAbs has been the conjugation of toxins or radionucleotides to further enhance their cytotoxic action. Other advancements that have occurred in the market include the development in antibody fragments, such as Fab and single chain antibodies, which can be produced at lower costs compared to whole antibodies.
Timeline of Antibody Development
| 1798 | Edward Jenner Demonstrates first smallpox vaccine |
| 1890 | Emil Von Behring show serum transfer from immunized animals could cure diphtheria |
| 1900 |
Paul Ehrlich proposed side chain theory, & models for antibody antigen binding & complement activation |
| 1901 | Emil Von Behring received Nobel Prize |
| 1948 | Astrid Fagraeus described role of plasma B cells in antibody generation |
| 1957 | Frank Burnet & David Talmage developed the clonal section theory |
| 1959 | Gerald Edelman & Rodney Porter published the molecular structure of antibodies |
| 1972 | Gerald Edelman & Rodney Porter received Nobel Prize |
| 1973 | First Atomis resolution structure of an antibody fragment published |
| 1975 | Georges Kohler & Cesar Milstein invent hybridoma technology |
Generation of Hybridomas
Production of monoclonal antibodies can be done in both in vivo or in vitro procedures or combinations thereof. The first step in these methods is the production of hybrid cells that are capable of producing the antibodies. Animals are generally used in the generation of mAb-producing cells. The final result of the the procedure yields a cell line capable of producing one type of antibody protein for a long period. Such an immortal cell line is called a hybridoma. Every hybridoma generation technique uses an animal. A technique which allows the intracellular production of antigen-binding antibody fragments, but this is done only in laboratory scale and their properties like yield, efficacy, and antibody function is uncertain.
Step 1: Immunization of Mice and Selection of Mouse Donors for Generation of Hybridoma Cells
Mice are immunized with an antigen that is prepared for injection either by emulsifying the antigen with an adjuvant or by homogenizing a gel slice that contains the antigen. Intact cells, whole membranes, and microorganisms are sometimes used as immunogens. In almost all laboratories, mice are used to produce the desired antibodies. In general, mice are immunized every 2-3 weeks but the immunization protocols vary among investigators. When a sufficient antibody titer is reached in serum, immunized mice are euthanized and the spleen removed to use as a source of cells for fusion with myeloma cells.
Step 2: Screening of Mice for Antibody Production
After several weeks of immunization, blood samples are obtained from mice for measurement of serum antibodies. Several humane techniques have been developed for collection of small volumes of blood from mice. Serum antibody titer is determined with various techniques, such as enzyme-linked immunosorbent assay (ELISA) and flow cytometry. If the antibody titer is high, cell fusion can be performed. If the titer is too low, mice can be boosted until an adequate response is achieved, as determined by repeated blood sampling. When the antibody titer is high enough, mice are commonly boosted by injecting antigen without adjuvant intraperitoneally or intravenously (via the tail
Step 3: Preparation of Myeloma Cells
Fusing antibody-producing spleen cells, which have a limited lifespan, with cells derived from an immortal tumor of lymphocytes (myeloma) results in a hybridoma that is capable of unlimited growth. Myeloma cells are immortalized cells that are cultured with 8-azaguanine to ensure their sensitivity to the hypoxanthine-aminopterin-thymidine (HAT) selection medium used after cell fusion. A week before cell fusion, myeloma cells are grown in 8-azaguanine. Cells must have high viability and rapid growth. The HAT medium allows only the fused cells to survive in culture.
Step 4: Fusion of Myeloma Cells with Immune Spleen Cells
Single spleen cells from the immunized mouse are fused with the previously prepared myeloma cells. Fusion is accomplished by co-centrifuging freshly harvested spleen cells and myeloma cells in polyethylene glycol, a substance that causes cell membranes to fuse. As noted in step 3, only fused cells will grow in the special selection medium. The cells are then distributed to 96 well plates containing feeder cells derived from saline peritoneal washes of mice. Feeder cells are believed to supply growth factors that promote the growth of the hybridoma cells. Commercial preparations that result from the collection of media supporting the growth of cultured cells and contain growth factors are available that can be used in lieu of mouse-derived feeder cells. It is also possible to use murine bone marrow-derived macrophages as feeder cells.
Step 5: Cloning of Hybridoma Cell Lines by “Limiting Dilution” or Expansion and Stabilization of Clones by Ascites Production
At this step new, small clusters of hybridoma cells from the 96 well plates can be grown in tissue culture followed by selection for antigen binding or grown by the mouse ascites method with cloning at a later time. Cloning by “limiting dilution” at this time ensures that a majority of wells each contain at most a single clone. Considerable judgment is necessary at this stage to select hybridomas capable of expansion versus a total loss of the cell fusion product due to underpopulation or inadequate in vitro growth at high dilution. In some instances, the secreted antibodies are toxic to fragile cells maintained in vitro. Optimizing the mouse ascites expansion method at this stage can save the cells. Also, it is the experience of many that a brief period of growth by the mouse ascites method produces cell lines that at later in vitro and in vivo stages show enhanced hardiness and optimal antibody production.
Advantages of In vitro Methods
- In vitro methods reduce the use of mice at the antibody-production stage (but can use mice as a source of feeder cells when antibody generation is underway).
- In vitro methods are usually the methods of choice for large-scale production by the pharmaceutical industry because of the ease of culture for production, compared with the use of animals, and because of economic considerations.
- In vitro methods avoid the need to submit animal protocols to IACUCs.
- In vitro methods avoid or decrease the need for laboratory personnel experienced in animal handling.
- In vitro methods using semipermeable-membrane-based systems produce mAb in concentrations often as high as those found in ascitic fluid and are free of mouse ascitic fluid contaminants.
Disadvantages of In vitro Methods
It should be noted that each of the items below pertains to only a fraction (3- 5%) of hybridomas, but they indicate some of the difficulties associated with in vitro methods.
- Some hybridomas do not grow well in culture or are lost in culture.
- In vitro methods generally require the use of FCS, which limits some antibody uses. The use of in vitro methods for mAb production generally requires the use of FCS, which is a concern from the animal-welfare perspective.
- The loss of proper glycosylation of the antibody (in contrast with in vivo production) might make the antibody product unsuitable for in vivo experiments because of increased immunogenicity, reduced binding affinity, changes in biologic functions, or accelerated clearance in vivo.
- In general, batch-culture supernatants contain less mAb (typically 0.002-0.01) per milliliter of a medium than the mouse ascites method. Note that semipermeable-membrane-based systems have been developed that can produce concentrations of mAb comparable with concentrations observed in mouse ascites fluid.
- In batch tissue-culture methods, mAb concentration tends to be low in the supernatant; this necessitates concentrating steps that can change antibody affinity, denature the antibody, and add time and expense. Adequate concentrations of mAb might be obtained in semipermeable-membrane-based systems.
- Most batches of mAb produced by membrane-based in vitro methods are contaminated with dead hybridoma cells and dead hybridoma-cell products, thus requiring early and expensive purification before the study.
- MAb produced in vitro might yield poorer binding affinity than those obtained by the ascites method.
- In vitro, culture methods are generally more expensive than the ascites method for small-scale or medium-scale production of mAb.
- The number of mAb produced by in vitro methods is limited by the amount of equipment that it is practical to have available.
- The Food and Drug Administration (FDA) estimates that proving the equivalence of an mAb produced by in vitro methods to an mAb previously produced by the mouse ascites method would cost higher.
Large scale production
In vivo production
The successful application of in-vitro or in vivo method depends on the biologic behavior of a hybridoma cell line. The concentration of mAb produced is also affected by the biologic behavior in case of ascites method Optimization of the production variables and selection of appropriate clones are done by the production facility personnel depending on their own application. For example, if cost is important in a particular application, system optimization often favors the economics of in vivo production. For cell lines that will go into continuous production, in vivo optimization is necessary and for cell lines to produce acceptable growth and mAb production, in vitro optimization is necessary. For in vitro production expense is an important factor. This production procedure requires high labor from highly paid, highly trained employees. OPtimization of disposable supplies is also required as it is an important factor in the increased costs associated with in vitro production.
In case of in vivo production variables like age, sex, the strain of the host, size of the hybridoma-cell inoculum, number of taps, and type and volume of primer should be optimized. The change in these factors affects the ascites yields and mAb concentration. The size of the tumor produced in the peritoneal cavity is indirectly proportional to the ascites produced. This leads to the selection criteria based on the size of the tumors. Sequential tapping provides the highest yields and greatest mAb concentration from a group of mice. This procedure has proved efficient except for very invasive cell lines that allow for only one needle tap. This tapping procedure also reduces the number of mice needed per gram of mAb by a factor of 2-3. The number of the taps should be maximized given the clinical condition of the mice is good. Generally, it should three taps. Optimizing the invasive nature of a cell line is needed to make sure the mice survive the completion of a production run. Selecting appropriate clones and altering hybridoma cell concentration injected into the peritoneal cavity of the mice are two ways to optimize production. The volume and concentration of mAb produced depend on the clone selected, and this makes systematic comparisons difficult. Therefore, the best way to achieve maximal in vivo yields is to screen clones in mice and to use the clone that provides the best yield. Cell growth conditions are optimal in vivo, so almost all cell lines will produce antibody, even when they are not optimized.
Once learned, Ascites production is a simple procedure. The optimal time for tapping the fluid is determined by skilled observers by daily observation of the mice. They also determine when the mouse should be euthanized. It is quicker and economical for small-scale and medium-scale production. It produces a higher concentration of mAb. Therefore it is easier to scale up in production. Purification costs of invivo methods. for most cell lines, is same as in vitro methods. The major problems associated with in vivo production are the use of animals, the possibility that the animal could be harmed if technicians are not properly trained and procedures are not followed properly, the presence of endogenous mouse immunoglobulin contamination except when immunodeficient mice are used and the possibility of contamination with murine pathogens, which requires the use of high-quality animals and a high-quality program for health assurance.
In vitro Production
Various in vitro commercial systems meet the different needs and requirements of users. These systems are of two types: single-compartment systems that allow only low-density cell culture and double-compartment systems that allow high-density cell culture, which results in increased mAb concentration.
For very-small-scale production (less than 10 g), the simple low-density cell-culture systems— such as culture flasks, roller bottles, gas permeable bags, and hollow-fiber bioreactors— are used. For small-scale and medium-scale production (10-100 g), double-compartment, high-density cell-culture systems, such as hollow-fiber systems, are used, as well as spinner flasks and roller bottles. High-scale production (over 100 g) is performed in large capital-intensive systems, such as homogeneous suspension culture in deep-tank stirred fermenters, perfusion-tank systems, airlift reactors, and continuous-culture systems.
An antigen-free product can be obtained by adapting the cell line to low-serum or serum-free media, with generally minor inhibitory effects on the cell line. Benefits of in vitro production are the absence of live-animal use, although some products in the culture media come from animals; the possibility of low-serum or serum-free media production; and the absence of host-contributed immunoglobulin or antigens. As the cost of disposable materials decreases further and technologic changes increase production efficiency and decrease equipment costs, the cost of in vitro production should decrease further, so it should become the preferred method of commercial production.
Problems associated with in vitro systems today are as follows material, labor, and equipment costs are higher than for the in vivo method; characteristics of the hybridoma are more critical than in vivo; about 3-5% of all clones cannot be maintained in existing in vitro systems; the great potential for microbial contamination, poor growth, and mechanical failure of the system or supporting systems requires constant monitoring and attention every day; production of large quantities of mAb is slower because of low mAb concentration, compared with the ascites method; the increased employee technical capabilities and educational background required by increased training time and system manipulations increase labor expense; the design of downstream processing is emphasized because large volumes of media are required to obtain large quantities of mAb and to ensure product economy and purity; and residual endotoxin, residual DNA from cell death, and bovine IgG contamination with cell lines that require some serum all complicate the process.
One of the most common causes of failure of in vitro methods is poor adherence to basic tissue-culture techniques, such as sterilization of culture-ware, equipment, and media and humidity and temperature control in the system. In large-scale and medium-scale production, it is important to have tight procedural and environmental controls to minimize losses due to system microbial contamination. To help avoid a major economic effect of such losses in commercial production, expensive facilities and tightly controlled procedures are implemented, all of which add to the high fixed cost of in vitro mAb production.
Some problems associated with Monoclonal antibodies
A Monoclonal Antibody May Be Too Specific
Binding of polyclonal antibodies to antigen gets effected only a little, due to small changes in the structure of the antigen owing to genetic polymorphism, heterogeneity of glycosylation or slight denaturation. The specificity of conventional polyclonal antisera depends on a consensus of hundreds of thousands of clonal products. They bind to antigenic determinants covering most or all of the external surface of the antigen. Hence, a subset of antibodies from a polyclonal antiserum will usually bind to antigens which have been modified or denatured. In contrast, monoclonal antibodies usually bind to one single unique site on the antigen molecule. In case of alteration in the binding site, the antibody may or may not continue to bind. Whether this is seen as a problem or an advantage will obviously depend on the individual circumstances. If the monoclonal antibody is used in a radioimmunoassay for a human serum protein, a minor genetic polymorphism (known or unknown) in that protein could cause gross errors. Similarly, if monoclonal antibodies were used for classification of microorganisms, they might not give exactly the same reactivity patterns as conventional antisera. It is therefore essential that monoclonal antibodies be tested and characterized in the assay system in which they are to be used. In future years, one may anticipate that commercial preparations of anti- bodies might be comprised of pools of several different clonal products, such that their nominal specificity will be maintained in all circumstances.
Affinity of monoclonal antibodies
For most of the polyclonal antisera, antibodies of varying dissociation constants from about 10– 6 M to 10– 9 M are present. The higher affinity antibodies tend to dominate in most practical situations and antibodies with affinities of less than about 10– 6 M are not usually detected by the standard methods. However, very high-affinity antibodies may comprise only a small fraction of the total. According to homogeneity of monoclonal antibodies means that each clonal product will have a well-defined affinity. The use of antibodies for affinity chromatography or immunofluorescence usually involves extensive washing to remove unbound antibody. If a monoclonal antibody has a low affinity, excessive washing may dissociate the antigen-antibody complexes. The extreme heterogeneity of polyclonal antibodies has a number of other important practical consequences. It allows an antibody population to be covalently modified (e.g. with fluorochromes or t25I). Although some antibodies will no longer bind, enough reactivity will be preserved to allow the experiment to proceed. Binding may still be detected even if more than 95% of the antibodies are destroyed. Monoclonal antibodies may behave quite differently under these conditions. In some cases, it may be possible to alter antigen or antibody quite extensively without destruction of binding. In others, it may be found that seemingly minor modifications to antibody or antigen may abolish binding completely.
Individual monoclonal antibodies vary greatly in the kinetics of binding to antigen. The binding of individual monoclonal antibodies to the cell surface may reach saturation in as little as 15 min, or as long as 90 min. It seems clear that the rate of association is sometimes limited by more than simple diffusion. These results might be understood in terms of the need for an individual antigenic determinant that is recognized by a given monoclonal antibody to be in a particular (transient) conformation before the antibody can bind. Such slow ‘on’ rates might be expected to be highly dependent on temperature. The highly individual kinetics of binding of monoclonal antibodies to their antigen makes it strongly advisable to keep incubation times constant from one experiment to another. Failure to do so may lead to errors in quantification and poor reproducibility in cases where the kinetics of binding are slow.
Purification
Primary Recovery Process
The first unit operation in a downstream process is the removal of cells and cell debris from the culture broth and clarification of the cell culture supernatant that contains the antibody product. The current followed trend for cell culture processes is to increase product titer through using enriched culture media, improving cell productivity and increasing cell mass. Although this may lead to a significant drop in cell viability. Those factors lead to an increase in the levels of process impurities such as host cell proteins, nucleic acids, lipids, colloids and the generation of a broad particle size distribution in the cell culture fluid (CCF).
Tangential flow microfiltration
The CCF flows tangentially to the microporous membrane and pressure-driven filtrate flow separates the soluble product from the larger, insoluble cells. Membrane fouling is limited by the inertial lift and shear-induced diffusion generated by the laminar flow across the membrane surface.
A high yielding harvest is achieved by a series of concentration and diafiltration steps. In the former, the volume of the CCF is reduced, thereby concentrating the solid mass. The diafiltration step then washes the product from the concentrated CCF mixture. Ideally, a pore size of 0.22 μm is employed for the TFF membrane as this will produce the target quality HCCF without the need for further clarification.
Centrifugation
Disk-stack continuous centrifuges are capable of removing cells and large cell debris; however, cells can be disrupted during the process, especially when the feedstock is a low cell viability culture fluid. Many particles of submicron size cannot be removed in the centrifuge, increasing the burden on the subsequent depth filtration.
The clarification efficiency of the centrifugation process is affected by harvest parameters such as centrifuge feed rate, G-force, bowl geometry, operating pressures, discharge frequency and ancillary equipment used in the transfer of cell culture fluid to the centrifuge. The cell culture process characteristics such as peak cell density, total cell density and culture viability during the culture process and at harvest will also affect separation performance.
Depth filtration
Depth filters are typically composed of cellulose, a porous filter-aid such as diatomaceous earth and an ionic charged resin binder. A binding resin is often added to a small weight percent to covalently bind dissimilar construction materials together, giving the resultant media wet strength and conferring a positive charge on the media surfaces. Because of this makeup, depth filters rely on both size exclusion and adsorptive binding to effect separation. Considerably thicker than a membrane filter, depth filters are approximately 2–4 mm in thickness. Depth filters are usually given a nominal pore-size rating, but these filters are far from absolute with regard to their particle size retention
For harvesting applications, depth filters can be applied directly to the whole cell broth or in conjunction with a primary separator such as TFF or centrifugation. The whole-cell broth depth filter harvest is common for bench, pilot and smaller commercial-scale applications. For this, a filtration train containing three stages of filters is usually employed. The primary stage uses a coarse or open, depth filter with a pore size range of up to 10 μm and removes whole cells and large particles. The secondary stage uses a tighter depth filter and clears colloidal and submicron particles. The last stage contains a membrane filter that is 0.2 μm pore size in most cases. The filtration process generally scales linearly; however, to ensure adequate filter capacity, a safety factor of 1.5X to >3X can be employed for each stage. Having too much excess capacity leads to large filter housing hold-up volumes, increased preparation time and floor space requirements and, ultimately, increased product loss due to hold-up.
Flocculation/precipitation
Flocculation of animal cells in suspension culture and selective flocculation of cellular contaminants from soluble proteins using acidic or cationic polyelectrolytes has been used for some time. Polyelectrolytes normally work by adsorbing to a particle to create an oppositely charged patch on the surface. This patch can then adhere to a bare patch on an opposing particle surface due to electrostatic attraction. The bridging mechanism of polymer adsorption to the cell and cell debris is the electrostatic attraction in most cases. Strongly cationic polymers are more effective at flocculating cells, whereas neutral and anionic polymers are often ineffective.
Precipitation
Precipitation with Ammonium Sulfate
Precipitation of immunoglobulins by ammonium sulfate is gentle, effective and simple. While it is not possible to purify immunoglobulins to homogeneity by this method, it provides a substantial enrichment, and reduces the protein load on subsequent purification steps. It is one of the oldest and most useful methods of purification of immunoglobulins. It is based on the observation that they are precipitated by lower concentrations of ammonium sulfate than most other serum proteins. It is simple to perform, but there are a few technical points which can greatly influence the degree of purification obtained. Asmmonium sulfate should be added slowly in the form of a saturated aqueous solution, rather than as solid crystals. High local concentrations of ammonium sulfate cause unwanted precipitation of proteins such as albumin, and thus degrade the degree of purification. It is customary to perform ammonium sulfate precipitations at 4ºC although most antibodies can be fractionated at room temperature without adverse effects. The percentage of saturation changes slightly with temperature, but for practical purposes the effect is negligible. There are few very important points which must be considered while opting for this method. The serum to be fractionated should be placed in a beaker on a magnetic stirrer with slow stirring, as frothing denatures protein. Saturated ammonium sulfate is added dropwise, allowing each drop to disperse before the next is added. When the concentration of ammonium sulfate reaches about 20% of saturation, the serum begins to turn milky. Most immunoglobulins will be precipitated by 35-40% of saturation, although it may be necessary occasionally to use up to 50%. Higher concentrations do not increase the yield of immunoglobulins, but will cause increasing contamination by other proteins, especially transferrin and albumin. The suspension should be stirred for 15-30 min, and then centrifuged at 2000 g or preferably 10 000 g. The supernatant should be clear, but may contain a layer of lipoprotein on the top. The pellet should be washed 2-3 times in 50% saturated ammonium sulfate. Washing the pellet considerably reduces contamination with nonimmunoglobulin proteins and does not lower the yield significantly. It should be noted that ammonium sulfate solutions are dense and resist mixing. Care must be taken that the stock solutions are adequately mixed. A few end-over-end mixings are desirable, as the mixing by magnetic flea may fail to disperse a dense lower layer. Solutions of saturated ammonium sulfate should not be stored in containers that were previously used for laboratory wash-up detergents. Finally, the precipitate is dissolved in PBS. It will virtually always redissolve easily. Ammonium ions interferes with many subsequent procedures, so it must be removed by overnight dialysis against 500-1000 volumes of compatible buffer. Ammonium ions interfere especially with conjugation of antibodies with fluorochromes, biotin or agarose. The dialysis fluid should be changed several times at intervals of a few hours.
In the early days of serum fractionation, it was noted that some serum proteins were soluble in distilled water, while others were not. The soluble fraction was known as albumin and the insoluble fraction as globulin. Subsequently, it was found that many globulin-like proteins were soluble in distilled water. These proteins were termed pseudoglobulins, to distinguish them from the water- insoluble euglobulins. Subsequently, the definition of globulins was broadened to include proteins precipitated by 50% saturated ammonium sulfate. Later still, the globulins were redefined to include any protein whose anodic mobility on electrophoresis was less than that of albumin. The term pseudoglobulin is now obsolete, but the term euglobulin persists. The euglobulin fraction of serum is composed mainly of a subpopulation of immunoglobulins, notably the majority of IgM molecules and a significant fraction of IgG and other classes. Precipitation of immunoglobulins at low ionic strength (euglobulin precipitation) is still occasionally used as a preliminary purification method. Some monoclonal antibodies are insoluble at low ionic strength, while others are not. Euglobulin properties are common among mouse IgM, IgG2b and IgG3 proteins, but rare in IgG 1. Euglobulin precipitation is usually carried out by dialysing the protein against distilled water. The pH should be about 6-7 because, like all proteins, immunoglobulins are least soluble near their isoelectric point. If necessary, low concentrations (c5 raM) of citrate-phosphate buffer, pH 5.8, may be used. Dialysis should proceed at 4~ for 24-96 h, with several changes. The resulting precipitate is harvested by centrifugation, washed twice in the dialy- sis fluid, and resuspended in isotonic PBS. One disadvantage of euglobulin precipitation is that it is sometimes difficult to redissolve the precipitate. Addition of NaCI to a final concentration of 0.3-0.5 M may help. According to Garvey et al. (1977), euglobulins precipitate more readily when dialysed against 2% boric acid (pH -~6), and the boric acid precipitate is more readily solubilized than other precipitates. The effectiveness of the boric acid is said to derive from its low ionic strength (it ionizes only weakly) and the fact that its pH is close to the isoelectric point of many immunoglobulins, especially IgM. In addition, the complexing of borate with the carbohydrate moieties on glyco- proteins may aid precipitation. The boric acid-glycoprotein complexes that are insoluble at low ionic strength are dissociable by higher salt concentrations.
Chromatographic Processes
Affinity chromatography
Affinity separation separates proteins on the basis of a reversible interaction between a protein and a specific ligand covalently coupled to a chromatography matrix. The technique makes for an ideal capture step in purification processes.
The high affinity of Protein A for the Fc region of IgG-type antibodies forms the basis for the purification of IgG, IgG fragments, and subclasses. The procedure typically employed for Protein A chromatography involves passage of clarified cell culture supernatant over the column at pH 6–8, under which conditions the antibodies bind and unwanted components such as host cell proteins and cell culture media components and putative viruses flow through the column. An optional intermediate wash step may be carried out to remove non-specifically bound impurities from the column, followed by elution of the product at pH 2.5–4.
Ion exchange chromatography
Separation by this technique is fairly selective, and the resins used are relatively inexpensive, so it can be applied early or late in a purification process. For an antibody having a basic isoelectric point (pI), cation exchange chromatography can even be used as an initial capture step, but most frequently ion exchange chromatography is applied as a polishing step(s) after the Protein A step. Ion exchange chromatography is ideal for reducing high molecular weight aggregate, charge-variants, residual DNA, and host cell protein leached Protein A and viral particles.
Anion exchange chromatography
Anion exchange chromatography uses a positively charged group (weakly basic such as diethylamino ethyl, DEAE or dimethylamino ethyl, DMAE; or strongly basic such as quaternary aminoethyl, Q or trimethylammonium ethyl, TMAE or quaternary aminoethyl, QAE) immobilized to the resin. It is a powerful tool to remove process-related impurities such as host cell proteins, DNA, endotoxin and leached Protein A, product-related impurities such as dimer/aggregate, endogenous retrovirus and adventitious viruses such as parvovirus, pseudorabies virus. It can be used either in flow-through mode or in a bind and elute mode, depending on the pI of the antibody and impurities to be removed. For antibodies having a pI above 7.5, which includes most humanized IgG1 and IgG2 antibodies, a flow-through mode can be a better choice to remove impurities. In flow-through mode, the impurities bind to the resin and the product of interest flows through. The column loading capacity, i.e., mass of antibody to mass of resin, can be quite high since the binding sites on the resin are occupied only by the impurities. For antibodies having a pI in the acidic to neutral range, which includes most humanized IgG4s, bind and elute mode can be used to remove process-related and product-related impurities from the product of interest.
Flow-through mode. Anion exchange chromatography in flow-through mode has been widely used as a polishing step in mAb purification processes designed with two or three unit operations to remove residual impurities such as host cell protein, DNA, leached Protein A and a variety of viruses. The operating pH is normally 8 to 8.2, with a conductivity of up to 10 mS/cm in the product load and equilibration and wash buffers. Conditions are chosen such that the product does not bind to the column, while acidic impurities such as nucleic acid and host cell proteins do. Depending on the resin, loading conditions and charge variant profile of the antibody product, the amount of product loaded can reach one hundred grams per liter of resin without compromising product quality. In general, the amount of product loaded in a flow-through mode depends on the impurity species and levels to be removed. A lower level of impurity in the product will result in a higher amount of product loaded.
Bind-and-elute mode
Anion exchange chromatography in bind-and-elute mode has also been implemented in the manufacturing of mAbs. The antibody product pool is first loaded onto an anion exchange column and the product of interest is then eluted with a higher salt concentration in a step or linear gradient, leaving the majority of impurities bound to the column. The impurities are eluted from the column during the cleaning or regeneration step.
In the development and optimization of an anion exchange chromatography in bind-and-elute mode, the operating pH should be above or close to the pI of the product in order to obtain a net negative charge or higher negative charge number on the surface of the antibody molecules, and hence to achieve a higher binding capacity during the chromatography step.
Similarly, to reach a high binding capacity, the ionic strength of the load should be in the low range and the pH should be less than pH 9. Manufacturing issues around the need to achieve lower ionic strength in the load will come into play, including the capability or need for diafiltration, product holding vessel volumes if the load will be diluted manually, and operational lower limits of buffer pump when running a gradient if the sample is to be diluted in-line.
Optimal elution conditions can be achieved using either a linear gradient or step elution. Step elution can deliver smaller product pool volumes and higher product concentration, while linear gradient elution can provide better process control, process monitoring, and reproducibility.
Cation exchange chromatography. Cation exchange chromatography uses a resin modified with negatively charged functional groups. They can be strong acidic ligands such as sulfopropyl, sulfoethyl and sulfoisobutyl groups or weak acidic ligand such as carboxyl group. Cation exchange chromatography has been applied to purification processes for many mAbs with pI values ranging from neutral to basic. Most humanized IgG1 and IgG2 subclasses are perfect candidates for cation exchange chromatography, in which the antibody is bound onto the resin during the loading step and eluted through either increasing conductivity or increasing pH in the elution buffer. The most negatively charged process-related impurities such as DNA, some host cell protein, leached Protein A and endotoxin are removed in the load and wash fraction. Cation exchange chromatography can also provide separation power to reduce antibody variants from the target antibody product such as deamidated products, oxidized species and N-terminal truncated forms, as well as high molecular weight species.
The dynamic binding capacity of mAbs on cation exchange resins depends on pH and conductivity. It is possible that protein molecules adsorbed at pore channels near the external surface of the media hinder other molecules from entering the pores and such exclusion is reduced at high conductivity, leading to a direct relationship between net protein charge and the solution conductivity yielding optimal dynamic capacity.
The maximum binding capacity attained can be as high as >100 g/L of resin volume depending on the loading conditions, resin ligand and density, but impurity removal depends highly on the loading density. High loading on the resin normally results in higher levels of impurities in the elution pool, although different ligands and resin bead sizes can have significant effects on the resolution of impurities. Therefore, a resin screening study should be performed to select the best resin, i.e., one with a high binding capacity and the best selectivity and resolution. The resin screening study is also linked with the elution condition development. The same principles described for anion exchange chromatography regarding development of the elution program apply to cation exchange chromatography as well.
Gel Filtration
Gel filtration separates proteins according to their size. The procedure is simple to perform and is capable of good recoveries. However, the process is slow, cumbersome and always results in dilution of the sample. Due to the sample getting diluted it is recommended to perform it before ion exchange chromatography, so that the ion exchanger can be used to reconcentrate the antibody with minimal losses. There is usually a much smaller factor of purification than ion exchange or affinity chromatography. The main role of gel filtration in purification of IgG is an adjunct to other methods, if a higher degree of purification is needed. Gel filtration has a very important role in the purification of IgM. Gel filtration is usually carried out in long, thin columns (typically 0.5-1 m long and 15-30 mm diameter). The gel itself occupies about 70% of the volume of the column, and the space between the beads the remaining 30%.
Membrane and Filtration Technology
Membrane and filtration technologies are used extensively in the isolation and purification of mAb and other recombinant DNA products, from the initial clarification of cell culture broth to the final sterile filtration of purified bulk solutions.
Membrane chromatography. Membrane chromatography or membrane adsorbers, function similarly to packed chromatography columns, but in the format of conventional filtration modules. Membrane chromatography uses microporous membranes, usually in multiple layers that contain functional ligands attached to the internal pore surface throughout the membrane structure.
In membrane chromatography, membranes consist of a polymeric substrate to which a functional ligand is chemically coupled. The polymer substrate is composed of multilayers of polyethersulfone, polyvinylidene fluoride and regenerated cellulose membrane. The most widely used functional ligands are the same as those used in chromatography resins, including ion exchange, e.g., quaternary amine (Q), diethylamine (D), polyethyleneimine (E), sulfonic acid (S) and carboxylic acid (C); affinity; reverse-phase, hydrophobic interaction and anionic mixed mode that is made of cross-linked Poly(Allylamine) (PAA).
Ultrafiltration
Ultrafiltration is a pressure-driven membrane process that is widely used for protein concentration and buffer exchange. Ultrafiltration is a size-based separation, where species larger than the membrane pores are retained and smaller species pass through freely. Separation in ultrafiltration is achieved through differences in the filtration rates of different components across the membrane under a given pressure driving force. Buffer exchange is achieved using a diafiltration mode in which buffer of the final desired composition is added to the retentate system at the same rate in which filtrate is removed, thus maintaining a constant retentate volume.
Ultrafiltration with membrane pores ranging from 1 to 20 nm can provide separation of species ranging in molecular weight from 500 daltons to 1,000 kilodaltons. This distinguishes the process from reverse osmosis, which involves membrane pores of less than 1 nm that allows water but not salts to pass, and from microfiltration, which has membrane pores ranging in size from 0.05 to 10 μm that allow proteins to pass through.
Ultrafiltration membranes have a unique “skinned” or anisotropic structure. Membrane selectivity is controlled by a thin skin layer that is approximately 0.5 μm thick, and the mechanical strength is provided by a thicker macroporous substrate. This allows retention to occur on the membrane surface rather than within the filter structure. Filtration fluxes are also greatly increased since a majority of the flow resistance takes place in the thin skin layer.
Viral Clearance
In the case of virus removal by low pH inactivation, critical parameters that should be monitored at bench scale are pH, temperature, and contact time. Solution composition and protein concentration also play important roles in process characterization. Low pH inactivation is very robust at scale-up and, unlike other downstream steps, once it is characterized at the small scale, it has minimal variation at the industrial scale. Scale-down ratios for virus filtration of 1 to ≥2000 can be achieved. Integrity tests must be performed and compared with virus removal performance; manufacturers usually provide this type of correlation. Scale-down virus-retaining filtration devices may have feed flow geometries that are not consistent with VLS production. As in the case of UF modules, shear stress due to air–liquid interfaces may be a problem. Minimum denaturation and aggregation should be achieved while varying feed flow parameters. As with UF, all scale systems are required to process the same volume-to-surface area, volume concentration factor, and DF wash volume.
Novel methods for monoclonal Antibodies production
A reactivation strategy has been developed in the generation of fully human mAbs. The technology employs in vitro culturing of immunized, specially-selected splenocytes with cytokines and low levels of antigen (Ag). This reactivation process yields affinity-matured, class-switched B cells.
The Advantages of this method are:
The mAbs are fully human, having been developed from human immune cells.
The approach does not require immunizing patients or access to convalescent blood, therefore, the platform is amenable to mAb development using de novo immunization against any therapeutic target.
mAbs developed using this approach will likely have higher affinities than Abs generated by library-based methods because reactivation focuses on the selection of affinity-matured cell targets that proliferate in very low Ag levels.
hu-mAbs generated using this process can have fully-human glycosylation patterns, a distinct advantage over human mAbs expressed in non-human cells.
The process enables selection for or against certain secondary characteristics such as Ab-dependent cellular cytotoxicity or complement-dependent cytotoxicity.
This process can be divided into four stages: immunization, reactivation, cloning, and expression.
Immunization
De novo immunization of human peripheral blood lymphocytes (PBL) in a SCID mouse model. The SCID-huPBL model repopulates immune deficient mice with human peripheral blood lymphocytes. SCID mice cannot make mature, antigen-specific B and T cells. Engraftment of human PBLs repopulates secondary lymphoid organs such as lymph nodes and spleen with human immune cells. Because of the enrichment process used, cells recovered from the immunized SCID-huPBL mouse are human, and the Ig genes cloned out from these cells are therefore fully human. All antigens are assayed for optimal concentration using a NanoDrop 8000 spectrophotometer before immunization. Antigen integrity and concentration are confirmed by gel electrophoresis and densitometry. Antigens are required to pass quality control by all three methods before they are formulated for immunization.
Reactivation
B cell culture and affinity screening. Activated B cells are enriched from the spleen and lymph nodes of immunized SCID-huPBL mice and then B cells are cultured in vitro with a mixture of cytokines. Antigen supernatants from the cultured B cells are tested for affinity and specificity using an Octet QK which uses BioLayer interferometry to determine affinity binding constants (KD) of purified Abs, as well as rank-order crude tissue culture supernatants by Kd estimates. Antigens are biotinylated and immobilized on a streptavidin Octet sensor using attachment methods recommended by the manufacturer. Reactivated culture supernatants are rank-ordered by affinity using crude supernatant samples. Generally, affinities from 10-9 to 10-11 M are considered an acceptable affinity range. Harvested cell cultures are then subjected to fluorescence-activated cell sorting (FACS) analysis in which single B cells specific for the immunizing antigen is deposited in one cell per spot on a 48-spot AmpliGrid glass slide.
Cloning
Recovery of Ig heavy and light chains, cloning, and sequencing. Single-cell reverse transcriptase-polymerase chain reaction (RT-PCR) is performed on an AmpliGrid system (Beckman Coulter) to capture the matched Ig heavy and light chain pairs from sorted cells. Ig heavy and light chains are cloned into antibody expression vectors and sequenced.
Expression
Protein expression and scale up. A NanoDrop 8000 spectrophotometer is used to quantitate purified preps of candidate hu-mAbs. Midiprep DNAs of paired Ig heavy and light chains cloned into antibody expression vectors are co-transfected into non-adherent mammalian cells using the TransIT reagent. Supernatants are screened for affinity and specificity as described above. Larger scale transfections can be performed on candidate hu-mAbs. Hu-mAbs can be purified by standard protocols and quantitated using the NanoDrop 8000 spectrophotometer for concentration and purity. Affinity constant (KD) determinations can be performed on purified antibodies. Additional assays to identify desired hu-mAb characteristics will be performed prior to stable cell line generation.
Recombinant Antibody (rAbs)
Since the discovery of hybridoma technology by George Köhler and Cesar Milstein in 1975 the targeted specificity of monoclonal antibodies have been used for number of applications as bio-therapeutics, diagnostic, medicine and research. The medical use of mAbs as human therapeutic agents have shown promising signs and there is increased demand for purified antibodies among the medical and scientific community. Various antibody engineering research have been carried out in the past three decades to enhance the quantitative and qualitative methods in antibody isolation, production and purification to meet the increasing antibody demand for diagnosis, therapeutics and research purpose.
With the recent advancement in recombinant DNA technology, monoclonal antibodies and binding fragments can be produced by gene modification techniques by enabling the combination of genetic material from one or more sources. In 1990 John McCafferty confirmed by conducting an experiment by inserting antibody DNA into phage genome using vectors, he research showed that that antibody fragments could be displayed on viruses that infect bacteria, called phages or aslo known as bacteriophages. Researchers then started to create libraries of antibody genes to display on phages and established procedures to successfully isolate individual antibodies from the large phage-displayed libraries. This method has become known as antibody phage display. These procedures were commercialized, thus enabling antibody production using recombinant method which offers a new convenient, efficient and rapid method for specific antibody fragments production for specific applications.
Recombinant antibodies involves using synthetic genes expressed in an in vitro mammalian cell line. This is a biologically and chemically defined animal-free system unlike traditional hybridoma based technologies which uses mice. Advances in molecular biology have lead to the ability to synthesize de novo antibodies in vitro completely without the use of animals.
General production methods for the manufacture of non-animal recombinant antibodies can be broken down into5 steps:
- Creation of an antibody gene library
Antibody gene libraries consists of collection of microorganisms that have been transformed with the genes encoding for the variable regions of different antibodies. A human phage display library is constructed by first isolating antibody RNA from a given source followed by ligation into a phage display vector. These vectors can then be used for expression of human IgG on bacteriophage hosts to represent the entire immune repertoire from which the RNA was isolated. One can then screen (or “pan”) a phage library for those which bind to a particular antigen and isolate the original IgG sequence.
Antibodies are Y-shaped molecule made up of heavy and light chains that each have a variable and a constant region. The variable region genes can either be synthesized in vitro or amplified from the genetic material in human antibody-producing B cells. Variable region genes are used instead of genes for whole antibody molecules because fragments of antibodies are more easily assembled in microorganisms than whole antibody molecules and the variable regions of an antibody are the most important fragments in terms of function. Each variable region gene is spliced into a vector (vehicles that transfer foreign genetic material into another cell), and the vector is taken up and integrated into the genome of a microorganism.
- Display of the library on Bacteriophage, phage or cell surfaces;
Antibody library vectors contain genetic instructions to produce a protein found on the surface of viral particles or cellular membranes. Genes encoding antigen-binding variable domains of antibodies are fused to phage genes that encode coat protein, allowing for the phage coat to express, or “display”, the antibody fusion protein when the vector is inserted into a microorganism. A collection of recombinant phage that display unique antigen binding domains on their surfaces is known as a phage display library.
- Isolation of antibodies against the antigen of interest
Once the recombinant antibodies are displayed, tools such as paramagnetic beads, fluorescence-activated cell sorting (FACS), and/or Enzyme-Linked Immunosorbent Assays (ELISAs) can be used to isolate individual antibodies that bind to a specific antigen target. Library members are incubated with the target molecule and unbound library members are washed away. The result is a library enriched for target-binders. This process can be repeated as needed to achieve the desired specificity. This process is called Exponential Evolution of Ligands by Systematic Enrichment or SELEX.
- Modification of the isolated antibodies
Bacteria, yeast or phage encoding the selected antibodies of interest are grown in greater quantities and are put through the selection process again to enrich for the strongest binding (highest specificity) candidates. If the affinities of the lead candidates are not strong enough, antibodies with higher specificity can be generated through “maturation” by random or rational mutagenesis.
- Antibody Expresssion:
Once an antibody is selected, the genes for that antibody are transferred into an expression system—bacteria, yeast, or mammalian cell lines specially designed for the expression of foreign proteins. The choice of vector and expression system depends on the type of antibody that is to be produced.
Antibody Heterogeneity
Monoclonal antibodies (MW 150 kDa) are composed of one heavy chain (MW 50 kDa) and two types of light chains (kappa and lambda, MW 25 kDa). From the point of initial production to the phase of elimination from the individuals system monoclonal antibodies goes through vrious modification, the antibody is changing over its lifespan and heterogeneity of monoclonal antibody is a common phenomenon. The vast number of modifications is a problem for thorough characterization of the molecules. Antibody can encounter various modification ranging from enzymatic to nonenzymatic modifications of monoclonal antibodies including the common ones such as incomplete disulfide bond formation, glycosylation, N-terminal pyroglutamine cyclization, C-terminal lysine processing, deamidation, isomerization, and oxidation, and less common ones such as modification of the N-terminal amino acids by maleuric acid and amidation of the C-terminal amino acid. In addition, weak associations with other molecules, conformational variety and aggregation of monoclonal antibodies also cause heterogeneity.
Monoclonal antibodies generally exhibit charge heterogeneity from oxidation, asparagine deamidation, aspartic isomerization, lysine truncation, glycan modifications, and other modifications. Therefore, the manufacture and testing procedures of MAbs involve routine analyses and monitoring of impurities resulting from these situations. Weak cation-exchange (WCX) columns are well suited and widely used for the characterization of MAb heterogeneity.
Applications of Monoclonal Antibodies
Antibodies, also known as immunoglobulins, are proteins in the blood that are created by B cells in response to proteins called antigens, which the body recognises as ‘non-self’. Understanding antibodies is useful because it means that we can develop blood tests to diagnose illnesses.
The four types of applications of monoclonal antibodies. The four types of applications are: (1) Diagnostic Applications (2) Therapeutic Applications (3) Protein Purification and (4) Miscellaneous Applications.
Diagnostic tests
When a patient comes to see a Doctor with a fever, muscle aches, a sore throat and a general sense of lethargy. The symptoms might suggest that the patient has contracted a viral or bacterial infection, to confirm and decide on the treatment, the Doctor will opt for serological analysis to confirm the presence or absence of bacterial/viral antigens. One way to tell the difference between the two, or any other pair of illnesses, is to look at your patient’s blood to see what type of antibodies they are producing. This should reveal which antigens are in their system. The entry of antigen can cause a number of different immunological reactions by the body, the evaluation of these immunological reactions like presence of IgM in the blood signifies an initial stage of infection, in some cases the immunological responses can be initiated by an allergic reaction causing histamines to be released by the mast cells. A thorough evaluation of these immunological responses can provide a wide range of diagnostic information. The presence of antibody against an infectious organism means the patient has had contact with that organism but not necessarily that he is suffering, or has suffered, from clinical infection. The changing levels of antibody, or analysis of the different components of antibody response, are usually able to provide clear information as to present or past infection.
Microorganism entry into the body, induces an immune response that involves cellular and humoral components. An immune response is usually characterized by antibody secretions. These can be measured in the laboratory through various biochemical and serological techniques. Most immunoassays rely on the formation of antibody- antigen complexes that can be identified using an indicator molecule.
A primary immune response occurs when a B cell sees an antigen for the first time. Antigen binding to the surface of the B cell stimulates the production of antibodies that are capable of binding directly to the antigen. The first recognition process takes time for antibody development and there is an initial delay for the body to fight the invading antigens. Immunoglobulin M (IgM) is an antibody produced during the primary immune response and plays a significant role in fighting the infection during this stage. Similarly when an antigen enters the system for the first time, large quantities of IgM are produced. Meanwhile, the B cells are producing highly specific Immunoglobulin G (IgG) more slowly. Once IgG is produced in sufficient numbers, the IgG plays a greater role in the removal of antigens from the body due to its ability to bind to the antigen specific binding accuracy. Increased numbers of circulating IgM can be seen in the bloodstream. The decrease of IgM is brought about as the amount of IgG increases in the blood stream. By measuring the changes in IgM and IgG a medical practioner will be able to identify the stage of infection and can decide on the doses and required therapeutical intervention.A ratio high in IgM indicates that an infection is in its early stages, while a ratio high in IgG indicates that the infection is in its later stage.
Under most circumstances the first component of an antibody response is IgM which rapidly declines as IgG antibody develops and this can persist for months or years. The IgG has a memory and can remember the same bacteria or microbes when it enters the body again, the IgG sends signal to B cell to produce more IgG and clears the antigen immediately before it can cause any infection.
Monoclonal antibodies have revolutionized the laboratory diagnosis of various diseases. For this purpose, MAbs may be employed as diagnostic reagents for biochemical analysis or as tools for diagnostic imaging of diseases. In general the assays are used:
- I) To evaluate the competence of the immune system itself
2) To measure a patient’s·immune response against foreign or selfproteins (antigens)
3) To identify components of hypersensitivity reactions producing disease.
Seroloical Analysis
Serology is the scientific study of blood serum and other bodily fluids. Serological immunoassays are designed to detect a particular antibody in patient serum samples, the analysis is dependent upon on standard and/or positive controls. Calibrators can be used in an immunoassay to produce standard curves to measure the test antibody concentration in the patient serum, or as positive controls to assess the performance of the assay and reagents. In practical immunological terms, serology is the diagnostic identification of antibodies in the serum. Serological tests are performed on blood serum, and body fluids such as semen and saliva. In practice, the term usually refers to the diagnostic identification of antibodies in the serum or the detection of antigens of infectious agents in serum. Such antibodies are typically formed in response to an infection (against a given microorganism), against other foreign proteins (in response, for example, to a mismatched blood transfusion), or to one’s own proteins (in instances of autoimmune disease).
MAbs in Biochemical Analysis
Diagnostic tests based on the use of MAbs as reagents are routinely used in radioimmunoassay (RIA) and enzyme-linked immunosorbent assays (ELISA) in the laboratory. These assays measure the circulating concentrations of hormones (insulin, human chorionic gonadotropin, growth hormone, progesterone, thyroxine, triiodothyronine, thyroid stimulating hormone, gastrin, renin), and several other tissue and cell products (blood group antigens, blood clotting factors, interferon’s, interleukins, histocompatibility antigens, tumor markers). In recent years, a number of diagnostic kits using MAbs have become commercially available. For instance, it is now possible to do the early diagnosis of the following conditions/diseases
Pregnancy
Pregnancy can be confirmed qualitatively by using a sensitive immunoassay kit with specific monoclonal antibody for human chorionic gonadotropin (hCG). Few ml of urine will be placed in the pregnancy kit well and allowed to react for few minutes, if there is presence of hCG in the urine it will bind to the mAbs to form a stripe which acts as a visual confirmatory test.
Hormonal disorders
Most of the hormones are present in very low quatity in oue blood stream, some as low as one pictogram(one millionth of a microgram) per mL. Due to its low concentration meseauring hormones in circulation blood proved difficultu in earlier. With the advent of radioimmunoassay the hormone can be detected in four steps.
Hormonal disorders analysis of thyroxine, triiodothyronine and thyroid stimulating hormone for thyroid disorders can be diagnosed by ELISA, radioimmunoassay and In Vitro methods
Infectious diseases
Infectious diseases by detecting the circulatory levels of antigens specific to the infectious agent e.g., antigens of Neisseria gonorrhoeae and herpes simplex virus for the diagnosis of sexually transmitted diseases.
MAbs in Diagnostic Imaging
Radiolabeled—MAbs are used in the diagnostic imaging of diseases, and this technique is referred to as immunoscintigraphy. The radioisotopes commonly used for labeling MAb are iodine—131 and technetium—99. The MAb tagged with radioisotope are injected intravenously into the patients.
These MAbs localize at specific sites (say a tumor) which can be detected by imaging the radioactivity. In recent years, single photon emission computed tomography (SPECT) cameras are used to give a more sensitive three dimensional appearance of the spots localized by radiolabeled— MAbs.
Immunoscintigraphy is a better diagnostic tool than the other imaging techniques such as CT scan, ultrasound scan and magnetic resonance. It is a nuclear medicine procedure used to find cancer cells in the body by injecting a radioactively labeled antibody, which binds predominantly to cancer cells and then scanning for concentrations of radioactive emissions. For instance, immunoscintigraphy can differentiate between cancerous and non-cancerous growth, since radiolabeled—MAbs are tumor specific. This is not possible with other imaging techniques. Monoclonal antibodies are successfully used in the diagnostic imaging of cardiovascular diseases, cancers and sites of bacterial infections.
Cardiovascular diseases
Myocardial infarction
Troponins are regulatory proteins in cardiac muscle that modulate the interaction between actin and myosin, during the calcium-mediated contraction of cardiac muscle. The absolute specificity of Troponin I for cardiac tissue makes it an ideal biomarker for myocardial injur. Troponin I test utilizes the principle of immunochromatography, with a unique two-site sandwich immunoassay on the membrane. If the intensity of the test band is equal to or greater than reference band, cardiac Troponin I (cTnI) concentration is equal to or greater than 1 ng/ml. The absence of colored band in the test region indicates a negative test result.
The cardiac protein myosin gets exposed wherever myocardial necrosis (death of cardiac cells) occurs. Antimyosin MAb labeled with radioisotope indium chloride is used for detecting myosin and thus the site of myocardial infarction. Imaging of radiolabeled MAb, is usually done after 24-48 hours of intravenous administration.
This is carried out either by planner gamma camera or single photon emission computed tomography (SPECT). It is possible to detect the location and the degree of damage to the heart by using radiolabeled antimyosin MAb. Thus, this technique is useful for the diagnosis of heart attacks.
Atherosclerosis
Thickening and loss of elasticity of arterial walls is referred to as atherosclerosis. Atherosclerotic plaques cause diseases of coronary and peripheral arteries. Atherosclerosis has been implicated in the development of heart diseases. MAb tagged with a radiolabel directed against activated platelets can be used to localize the atherosclerotic lesions by imaging technique.
Cancers
Cancer cells have undergone a series of genetic changes and therefore are likely to express novel
Or altered proteins known as tumor antigens. Some of the known tumor associated antigen are CA -15-3,CA 19-9, CA72-4, CA 125, CEA, α-fetoprotein (AFP). Besides diagnosis, estimation of tumor markers is also useful for the prognosis of cancers. That is a gradual fall in a specific tumor marker is observed with a reduction in tumor size, following treatment.
Monoclonal antibodies against many types of human cancers are now available. Tumors can be located in patients using radioisotope labeled MAbs specific to the protein(s), particularly of membrane origin.It has been possible to detect certain cancers at early stages (lung cancer, breast cancer, ovariran cancer, malanoma, colorectal cancer) by employing MAbs. About 80 per cent specificity has been achieved for detecting cancers by this approach.
Bacterial infections
In recent years, attempts are made to detect the sites of infections by using MAbs. This is made possible by directing MAb against bacterial antigens. Further, monoclonal antibodies against inflammatory leucocytes which accumulate at infection site are also useful to specifically detect localized infections. The immune system acts as defence against various infectious agents that cause different forms of diseases. Two major components are the humoral (antibody-mediated) and cellular (cell-mediated) immune responses. The humoral immune system which comprises B-lymphocytes recognizes the type of foreign invading antigens and produces specific antibodies against them. The two important characteristics of an antibody are its specificity to the antigen, and its assurance to provide continual resistance to that particular type of antigen. Considering their unique features, scientists use them for the protection of humans against diseases. Techniques for in vitro production of antibodies was also developed, resulting in the production of monoclonal antibodies for diagnostic and therapeutic applications.
Previously WIDAL test was the most widely used confirmatory test to check for presence of bacteria Salmonella typhii. The WIDAL test is time consuming, cumbersome, uses lot of reagent and requires a trained microbiologist to conduct the analysis. Recent advancement in antibody engineering has led t the development of In vitro Diagnostic (IVD) kits which has come as a boon to areas where typhoid is endemic. These IVD kits can be used by persons with minimal knowledge on basis science and the result can be obtained in 5 minutes without the use of extensive lab wares and reagents. Similar kits have been developed for detecting the presence or absence of microbial antigens such as malaria, dengue, chikenguniya & syphilis.
Therapeutic Treatment
Recent advances in genetic engineering have made possible efforts to improve the therapeutic application of mAbs by identifying new targets with improved efficacy for use in clinical practice. Their use in immunoprophylaxis or immunotherapeutics have been extensively applied to infectious diseases, as carriers for toxic substances delivery to tumors or as tools for identifying, locating and target neoplasms. Progress in antibody engineering has yielded various types of mAbs for application in life science and biomedicine. They have also been used in the treatment of several types of cancers, immune diseases, arthritis and metabolic diseases. These types of antibodies may have similar principles, but different targets and applications. In addition, the choice of one method over another may be guided by several factors, including purpose of application, availability and effectiveness. Their therapeutic applications include cancer therapy, human and animal disease therapy, preparation of vaccines, and suppression of immune response and purification of hormones. Antibodies also play an important part in the development of several diseases. They can render infectious organisms harmless by attaching to their antigens, and can act as bio-therapeutics.
Due to development of new phase of therapy in the field of medicine, application of mAbs in the treatment of several disease conditions have been at the forefront. In 2002, the first human mAb for use in clinical practice was approved by the Food and Drug Administration of the United States. Since then, the mAbs production industry has exponentially expanded. About 30 mAbs were recently accepted for clinical use as therapeutics, with several others been at various trial stages.
Monoclonal antibodies are routinely used in biochemistry, molecular biology and medical research but the greatest achievement has been their use as therapeutic agents for the treatment of diseases such as breast cancer, leukaemia, asthma, arthritis, psoriasis, Crohn’s disease and transplant rejection. Important advances have been made over the past decade to improve the engineering technologies, safety and efficacy of the first generation of therapeutic antibodies. These developments, along with a greater understanding of the immunomodulatory properties of antibodies, have paved the way for the next generation of new and improved antibody-based drugs for the treatment of human diseases.
Types of Therapeutic Monoclonal antibody
Murine Antibodies
Köhler and Milstein’s method, which is still useful today, involves fusion of a cancerous (immortal) mouse B-cell myeloma with an immunized mouse plasma cell, creating a hybrid cell, or hybridoma. Murine mAbs are entirely derived from mouse IgG and were the first type of mAbs to be developed. However, murine mAbs incur problems with immunogenicity, which mounts a response known as the Human Anti-Mouse Antibodies (HAMA) response, producing unwanted side effects. The use of murine antibodies produced by hybridoma technology in human therapy (clinical medicine) is limited, attributable to differences between the human and rodent immune systems. This usually results in treatment failure, with the exception of some particular circumstances. Murine antibodies have mild effects of stimulation of cytotoxicity. Thus, their continuous administration often results in allergic reactions and anaphylactic shock, a result of the production of human anti-mouse antibodies (HAMA) that invariably attack the administered murine mAb and in turn stimulate allergic response. Since murine mAbs contain foreign protein molecules, majority of the early reagents for clinical use stimulated unwanted immune responses in human patients.
Recently, molecular biology advances have brought about in vitro gene manipulation and subsequent expression of these manipulated sequences in mammalian, bacterial or fungal cell culture protocols. This has thus ensured a better option for re-engineering murine mAb to partly substitute the rodent antibody fragment with a corresponding human antibody sequence.
Chimeric Antibodies (cAb)
Chimeric antibodies are special types of therapeutic antibodies made by the combination of genetic ingredients from humans and nonhumans (mice, rabbit). They are produced through manipulation of human constant regions and mouse variable regions. The chimeric antibodies retain the original antibody’s antigen specificity and affinity and are valuable tools for in vivo and in vitro research. These antibodies are made up of about 65% human genetic component in order to minimize the risk of unwanted reactions to foreign antibodies. Chimeric mAbs have an improved immunogenic profile in comparison to murine mAbs, but have more side effects than humanized and fully human mAbs.
Many therapeutic monoclonal antibodies were originally generated in mice and chimerized to reduce immunogenicity in humans. Examples of chimeric antibodies in the medical use include infliximab, rituximab and abciximab. With the emergence of recombinant DNA technology, monoclonal antibodies with greater amounts of human sequences are now being developed and used. The first step in structuring the ideal human monoclonal antibody was the chimeric antibody. Chimeric antibodies are composed of protein sequences from two origins: murine and human. While murine antibodies are 100% murine protein, chimeric antibodies are typically only about 33% murine proteins with the remainder human protein. In chimeric antibodies, the variable region with the antigenic specificity remains murine. The constant regions which dictate the antibody isotype are replaced with human proteins. Chimeric antibodies are made by melding murine variable region genes with human constant region genes. One recombinant DNA method by which this can be accomplished is by isolating the variable region genes from a murine hybridoma secreting an antibody that binds the desired target, then amplifying these genes using polymerase chain reaction (PCR.) This initial product is a copy DNA (cDNA) of the murine variable region (V-cDNA) The V-cDNA can then be ligated into a plasmid (small circular, extra chromosomal DNA in certain bacteria). Monoclonal Antibody chimerization is an important and powerful tool to reduce immunogenicity when injected into a different species. Chimerization or humanization is also a must-do step to convert a mouse monoclonal antibody into a therapeutic antibody candidate for human use.
Humanized monoclonal antibodies (HMA)
A recombinant DNA containing the promoter, leader, and variable region sequences from a mouse antibody gene and the constant-region exons from a human antibody gene is cloned to produce an antibody. The antibody produces is called as humanized antibody. The specificity to the antigen is caused by the variable region is obtained from the mouse DNA, its isotope which is determined by the constant region is obtained from the human DNA. Humanized monoclonal antibodies typically retain only the hypervariable regions, or complementary determining regions (CDRs), of a murine antibody while the remainder of the antibody is human. Thus, humanized antibodies typically contain only 5% to 10% murine composition. In humanised antibodies, the hyper variable regions are grafted onto human variable domain framework. The antibody molecules is nearly 95% human origin.
Human mAbs (HMA) have been considered as a source of natural and safe drugs due to their in vivo activities. Reforms in the field of mAb technologies has made human mAbs to be widely applied in the therapy of various diseases, as well as in the development of novel immunodiagnostics. A number of about 20 mAb drugs, including humanized mice mAbs, have been accepted as therapeutic reagents during the past few decades. Other mAbs are in different stages of clinical trial, and controlled by different research institution, and/or in collaboration with pharmaceutical companies. Examples of FDA-approved humanised antibodies include daclizumab, omalizumab, alemtuzumab.
Human monoclonal antibodies
Human monoclonal antibodies are fully, or nearly 100%, human in composition. The word monoclonal is technically not applicable to all synthetic human antibodies because some of the synthesis technologies do not employ hybridoma technology.
There are two basic technologies employed to produce human antibodies:
(1) genetically engineered, knockout or transgenic mice; and
(2) use of phage display libraries.
Knockout mice are developed by harvesting embryonic stem cells from early stage fertilized mouse embryos. An existing gene is inactivated or “knocked out” by replacing it with an artificial piece of DNA. The altered stem cells are then grown into mice with an altered genomic profile. Inactivation of the ability to rearrange germ-line heavy and light chain configurations inhibits the mouse ability to make murine immunoglobulin and the corresponding murine B-cells. Substituting human heavy and light chain germ-line DNA can cause the knockout mouse to produce human B-cells which may produce human monoclonal antibodies. While technically challenging, the use of knockout mice to produce entirely human monoclonal antibodies is an emerging and promising technology.
Human mAb production by hybridoma techniques is fairly difficult because of the hassle involved in maintaining immortalised cell lines and human hybridomas. It is also not feasible for in vivo immunization of humans with many different antigens compared to the use of animal models. However, methods for the production of human mAbs are made possible through the expression of antibody fragments or single cell variable fragment (Fab or ScFv) in bacteria. Similarly, antibody fragments can be displayed on filamentous bacteriophages for screening of antibody libraries. Generation of fully human mAbs serves as an alternative to reengineer murine mAbs with a source of low immunogenic therapeutic antibodies.
Monoclonal T-cell Receptors (TCRs)
Antibodies can bind to molecules expressed at the surface of target cells (as well as to soluble molecules) but are not effective against the peptide fragments that antigen-presenting cells contain tucked within their histocompatibility molecules. T-cell receptors are the ligands needed for that job. So monoclonal antibodies are not effective against intracellular antigens, e.g. virus-encoded proteins and tumor-specific antigens. But now progress is being made toward the development of monoclonal T-cell receptors (αβ TCRs).
Two ways in which these molecules could be helpful:
- Transforming normal T cells — whatever their natural specificity — so they also express a new TCR of desired specificity (and high affinity). These could then be introduced into a cancer patient to target the tumor-specific antigens or into an AIDS patient to target HIV-infected cells.
- Preparing a fusion protein of
- the engineered TCR conjugated to
- an effector molecule to destroy cells expressing the target MHC-peptide complex.
Monoclonal antibodies future
The market for protein-based biotherapeutics is large and is still growing rapidly. In 2015, 27% (12/45) of the drugs approved by the Food and Drug Administration (FDA) were biologic products. The largest group of biologics are antibody-based, mainly mAbs. There are 61 antibody-based products currently approved and in review in Europe and the US (as of October 2016). Recently, biosimilar products have also entered the market. They are a biologic medicinal product that contains a highly similar but not identical version of the original active substance of an already authorized original biologic medicinal product. The European Medicines Agency (EMA) has approved 21 biosimilars to date, including the first mAb biosimilar of infliximab, Inflectra (infliximab-dyyb). In March 2015, the FDA approved its first biosimilar product (Zarxio™, filgrastim-sndz, Sandoz), which was followed by the first US mAb biosimilar approval, in April 2016 (for Inflectra). Use of animal in the development of mAbs, as well as other protein-based biotherapeutics and biosimilars, poses unique challenges. The scientific and regulatory community is focussing on continued evaluation and integration of emerging technologies to reduce and refine animal use for biotherapeutic mAb development. However, there are still several barriers that must be recognized and overcome to make this a reality. The individual case studies showed that the ability of existing in vitro and in silico technologies to detect or predict toxicities observed in in vivo studies was lacking. Therefore, the challenge for the future will be to advance and apply novel technologies that have the capability to more closely represent the in vivo situation. Due to the species specificity of many products, non-human primates (NHPs) have been used for nonclinical safety and toxicology testing. However, there are often fundamental differences between primate and human physiology. Due to which there are deficiencies in the translation of NHP study results to human. There can be different situations like, if the target does not play a role (or is redundant) in normal physiology in NHPs. There might be cases where the target is still present, but has a different role or downstream effect in primate. In such situations, studies in NHP may therefore be of limited value to human risk assessment. Alternatively, mouse target knockout phenotypes can be used for hazard identification, or surrogate molecules can be used in rodent species to demonstrate safety and efficacy. An argument for the unnecessarily increase NHP use in the future was an increase in juvenile toxicity studies as default practice to support mAb development in pediatric populations. It was due to regulatory perception within companies and previous requests from the Pediatric Committee (PDCO/EMA). To prevent unnecessary conduct of these studies as the general rule, a data-sharing initiative will be needed to evaluate whether juvenile toxicity studies in animals provide any additional clinically relevant information, and if so, in what circumstances.
Toxicological science is also advancing rapidly across the public and private sectors. There is regulatory interest in these approaches nationally and internationally at the Organization for Economic Co-operation and Development level (OECD). In the chemicals industry adverse outcome pathways (AOPs), data from the USA’s ToxCast program and exposure-based modeling are being applied to human risk assessment. So, the focus is toward human-specific mechanisms of action and pathways-based approaches. Many of these approaches, in combination with technologies emerging from the research base, also show the potential to transform the pharmaceutical industry. The availability of new technologies alongside recent publications that have questioned whether the use of NHPs adds scientific value to the development of mAbs suggests that the timing is right to review the current biotherapeutic mAb development paradigm.
Since 2014, FDA has approved at least five monoclonal antibodies per year. These therapies encompass a number of clinical indications. Despite their promise, monoclonal antibodies have not had a smooth road to approval. Over the years, even seemingly well-designed monoclonal antibodies have often resulted in unacceptable adverse reactions. Hence a need is there to devise new ways to make the technology viable. The big breakthrough that pushed monoclonal antibodies from a good idea to a clinically useful tool came about with the advent of antibody humanization.
For future biotherapeutic mAb development a number of areas have been identified for future. These include the development and increased use of emerging technologies such as cell and tissue-based technologies that are suitable for mAb development, and opportunities to waive chronic and juvenile toxicity studies. Currently, many of the emerging technologies are being developed with small molecule new molecular entities in mind, rather than biologics. A number of organizations have an interest in progressing this area across the regulatory, industry and public sectors.
The majority of clinically relevant findings for mAbs are based on their mechanism of action. However, the toxicities presented in the case studies were, in general, not predictable before in vivo studies despite their relationship to the pharmacological action of the mAb. It is also important to note that many associated clinical toxicities such as some cancers, progressive multifocal leukoencephalopathy and infection, are so rare that they are not realistically detected in any in vitro or in vivo study. Currently, the field remains insufficiently confident in the ability of in vitro models to capture unpredictable toxicological findings as highlighted in the case studies. The development of more sophisticated and relevant in vitro technologies for safety assessment of mAbs may need to be more case-dependent, to take in to account their innate complexity, diversity and size, as well as their specific mechanism of action.
A future vision for mAb development is one in which fewer animals are used, but where the data obtained are more predictive of human safety. Therefore the typical approach to safety assessment of mAbs was considered. Typically two studies, one to support FIH clinical studies (IND-enabling) and one to support registration, are performed during mAb development. In some cases for oncology indications, a single study may suffice. However, an alternate approach could be to use a single, comprehensive in vivo study for the majority of mAbs that includes, in addition to toxicity endpoints, relevant pharmacodynamic, biomarker and potentially safety pharmacology endpoints. It has been argued that new clinically relevant findings are rarely identified in long-term studies that were not observed or could not have been predicted from the short-term study. Future work will involve whether both the 12-week and 26-week studies are of value in detecting clinically relevant findings. Of course, it is often the rare cases that ultimately drive regulations to ensure adequate safety in clinical trials. One recommendation for further progress in this area is to generate an evidence-base to help determine the frequency and types of toxicity that are observed only in long-term studies (6 months) compared with the shorter-term studies (1-3 months), and to explore whether these risks might be predicted in advance for specific types of targets.
For researchers working with monoclonal antibodies, supporting reagents and complementary tools are essential for effective experiments. ELISA kits can be used to quantify antibody binding and validate antigen recognition, while recombinant proteins provide highly specific targets for mAb testing. Researchers may also explore the use of polyclonal antibodies for broader antigen detection or employ secondary antibodies to amplify signals in various assays. Together, these resources enhance the reliability, reproducibility, and scope of monoclonal antibody research and applications in diagnostics, therapeutics, and biomedical studies.
It is difficult to predict what the future of antibody humanization will look like. Over time, the humanization process of mouse-derived antibodies has become much more sophisticated, with several companies now performing in silico optimization of CDRs for T-cell epitope avoidance to reduce immunogenicity. Yeast display is a new technology that in theory avoids some of the disadvantages of the phage display process for monoclonal antibody generation. Newer transgenic mice that have a fuller complement of human antibody genes are also being used to develop a new generation of fully human monoclonal antibodies. Regardless of the exact path that gives rise to the next generation of monoclonal antibody therapies, one thing is for certain: therapeutic antibodies can do things that few small molecules can, and as such will remain firmly a part of the drug development landscape for years to come. It is clear that there are opportunities to improve mAb development, but this will not happen without the collective knowledge, experience and dedication of experienced drug safety professionals and a more open-minded approach to mAb development.