Southern Blotting
Brief Introduction: Blotting Techniques
Blotting is used in molecular biology for the identification of proteins and nucleic acids and is widely used for diagnostic purposes. This technique immobilizes the molecule of interest on a support, which is a nitrocellulosic membrane or nylon. It uses hybridization techniques for the identification of the specific nucleic acids and genes. The blotting technique is a tool used in the identification of biomolecules such ad DNA, mRNA and protein during different stages of gene expression. Protein synthesis involves expression of a DNA segment which gets converted to mRNA to produce the respective protein. Molecules such as DNA, RNA and proteins are subjected to biochemistry analysis which are separated using blotting techniques. In the case of a cell, these molecules are present altogether and hence with the help of blotting scientists are able to recognise a specific molecule out of all others. Blotting is performed by allowing a mixture of molecules of interest pass through a block of gel which separates the molecules based on their molecular sizes. The hence processed molecules are required to be hard-pressed against a suitable membrane which will in turn transfer the molecules from the gel onto a suitable membrane (nylon, nitrocellulose or PVDF) via capillary action. After the molecules are transferred to the membrane their position does not change.
Southern blotting was introduced by Edwin Southern in 1975 as a method to detect specific sequences of DNA in DNA samples. The other blotting techniques emerged from this method have been termed as Northern (for RNA), Western (for proteins), Eastern (for post-translational protein modifications) and South-western (for DNA-protein interactions) blotting.
Subtypes of blotting such as northern, western & southern depend upon the target molecule that is being sought. When a DNA sequence is the foundation or code for a protein molecule, the particular DNA molecule of interest can be blotted using Southern Blotting technique. During gene expression, when the DNA is expressed as mRNA for a protein production, this process can be identified by Northern blotting. Finally, the coded mRNA produces the concerned protein, this protein identification can be done by Western Blotting.
Blotting approaches are viewed as an aide to the gel electrophoresis which is generally applied for separation of DNA/RNA/protein and yields reproducible results attributed to their excellent resolving power. Thereby, specific molecules can be detected amid the combination of molecules that are subjected to the separation. Most of the methods have a general step wherein the molecules of interest are transferred once separated are transferred from the gel to a solid membrane phase which is accomplished by drenching in a solution across the gel and the membrane via penetrable paper. Many types of multifaceted apparatus are also provided from a lot of suppliers for electroblotting which is more specifically useful for transfer initiated from less porous polyacrylamide gels compared to commonly used porous agarose gels. In case of DNA and RNA the detection of specific sequences in the membrane are carried out via hybridization with nucleic acid labeled probes which in the case of proteins is replaced by the use of labeled antibody probes. The initially developed protocols applied radioactive probes labeled with, radioactive isotopes for detection purposes via implementation of autoradiography procedures. In this process using the pattern of decay emissions radiated from a radioactive material is applied to produce an image on an x-ray film which can also be made available as a digital image by application of scintillation based gas detectors or systems based on phosphorimaging. Keeping in mind the harmful effects of exposure to radioactivity other kinds of labeling systems have been developed which includes fluorescent and chemiluminescent reagents. Luminescence/fluorescence based methods are considered to be sensitive and background free thus generates more accurate results. Also, there are several other modifications that have been implemented in contrast to the original method like DNA probes are more commonly applied rather than RNA, the nylon membranes have replaced the traditional nitrocellulose, to avoid the renaturation of DNA sequences during transfer the transfer is carried out in alkaline which was supposed to be in neutral solution as per the original protocol. The DNA is subjected to acid treatment for reducing its size inorder to increase the transfer rate of larger fragments. The gel strips and tube gels applied in the original protocol are no longer used, instead gel formats are applied. Though there have been many changes in the original protocol still the modern day protocols retain most of the fundamental features of the original protocol.
Southern blotting was applied in many important studies like the genetic mapping of the human genome which was based on blotting based detection of restriction fragment length polymorphisms. Also, the DNA fingerprinting was first developed via hybridization of the human DNA restriction digestion products with minisatellite probes. However, most of the primary applications of this method have been now substituted by DNA sequencing and polymerase chain reaction (PCR) as they provide more extensive facts or data and moreover are easy to implement. Nevertheless, blotting is still applied in a lot of arenas like in the measurement of copy number, analysis of long stretches of DNA that is difficult to be amplified or sequenced using PCR or DNA sequencing and in case of structural analysis of DNA wherein the physical forms of DNA are separated using two-dimensional gel electrophoresis and thereafter it is detected using blotting of specific components.
General Procedure for blotting
Homogenization of the sample which involves the purification of DNA/RNA/proteins which is performed after extraction from a variety of sources such as cells or tissue.
Digest the DNA with restriction enzymes into fragments, which is not required for RNA(northern blot).
Separation of the molecule of interest by an electrophoresis membrane generally on an agarose gel for DNA fragments. In the case of RNA samples they can be separated on an agarose gel in presence of formaldehyde as the denaturing agent. This is necessary as formaldehyde confines secondary structures of RNA molecules.
Transferring the molecules (DNA/RNA fragments) to a nitro cellulosic membrane/ nylon membrane from the gel.
Prehybridization (Blocking): Washing of the nylon membrane with a prehybridization or blocking solution comprising salmon sperm DNA is required inorder to block non-specific DNA interactions and also this helps in the reduction of background noise. As an alternative, there are some commercially available blocking buffers loke PerfectHyb™ Plus buffer in which there is no requirement of salmon sperm DNA for blocking purpose.
For the preparation of probe fresh probe DNA labeled with 32P alpha-labeled dCTP is prepared.
Hybridization or identification of the molecule which is achieved by incubating the blot with the specific labeled probe.
For the detection of the probe and the sequence of interest DNA/RNA the film is exposed to -80°C.
Southern Blotting
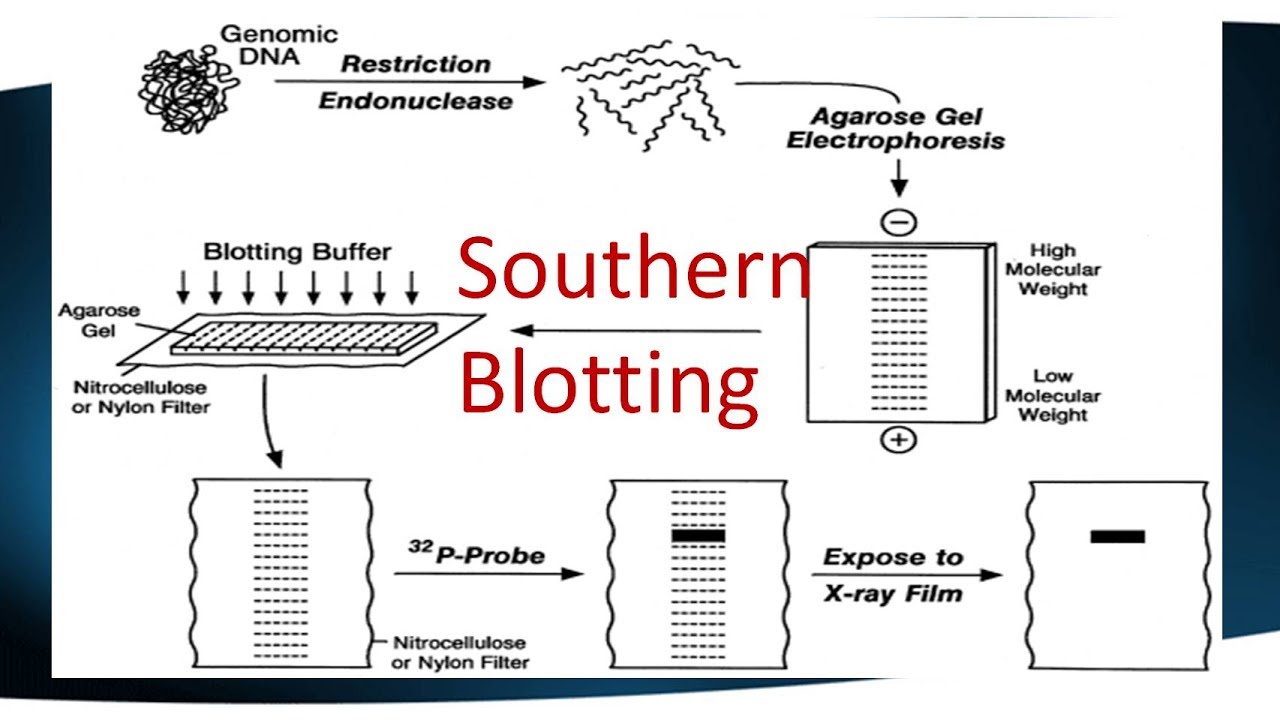
The first of these techniques developed was the Southern blot, named after Dr. Edwin Southern who developed it to identify specific DNA sequences. Southern blotting is a detection technique used to find the target DNA sequences in the DNA sample in the field of molecular biology. The process starts from electrophoresis of DNA molecules which are hybridized in a blotting membrane followed by a transfer step where DNA from gel is transferred onto the blotting membrane.
Historical background
The ability of the DNA strands to hybridise come from its complementary nature clearly depicted in Watson-Crick’s DNA structure model. In the beginning experiments were carried out to study the renaturing ability of DNA and formation of DNA/RNA hybrids for understanding tissue or cell specific expression. Thereafter hybridisation came into picture with the development of probes which can detect specific sequences within the target and immobilisation of the target sequences onto a solid support like nitrocellulose powder which further gave rise to nitrocellulose membranes. The demonstration by Southern that DNA fragments separated by gel electrophoresis can be transferred onto a membrane allowed characterisation of DNA by hybridisation pattern and by their molecular sizes. Later this method was applied for spatial identification of targets and blotting of bacterial colonies.
The electrophoresis step can be done using two different types of gels such as Polyacrylamide gel (PAGE) with urea and Sodium Dodecyl sulfate (SDS) with urea. These two gels have different application. When a PAGE gel is used, the same quantity of DNA fragments is transferred to the blotting paper. If a SDS is used the resolution of the bandwidth formed remains same. In this technique DNA molecule of size, 100 pg can be identified. The technique can be summarized as the formation of double stranded DNA which has one strand from target DNA and the other from DNA probe. DNA probe is produced in vitro for the sequence of interest.
Principle
Restriction endonucleases, which is an enzyme, is used to break the DNA into small fragments. These fragments are then separated using electrophoresis. The fragments achieved is then classified according to their size (kDa). Thus, DNA fragments are transferred to the blotting paper where it is incubated with probes. Probes used in the Southern blotting can be highly selective. They can selectively bind with a resolution of 1 in a million and the characteristics to bind to the intended target fragments.
Materials Required
Reagents
- The buffer used for electrophoresis is TAE or TBE.
- The agarose preferred should be of electrophoresis grade.
- For staining the DNA, ethidium bromide (0.5 µg ml–1, dissolved in H2O) is used. But there are other DNA staining dyes such as SYBR green which can replace ethidium bromide for safe handling.
Note: Ethidium bromide being a mutagen requires careful handling. It is advised to wear gloves during its usage and follow the pertinent regulations for disposal of pipette tips. Avoid touching any objects with ethidium bromide contaminated gloves.
- 2Xand 20X SSC (the composition of 20X SSC includes 3.0 M NaCl and 0.3 M sodium citrate)
- 6X DNA loading buffer composed of 0.25% bromophenol blue, 0.25% xylene cyanol FF and 30% glycerol in water.
- Suitable DNA markers of varying molecular weight also referred to as DNA ladders are used as standards for reference.
- The prehybridization and hybridization mixture consists of 0.5% SDS, 6X SSC, 5X Denhardt’s solution and 100 mg ml–1 sheared, denatured salmon sperm DNA or yeast tRNA.
- Denhardt’s solution widely used for hybridization is made up of 0.02% Ficoll, 0.02% polyvinylpyrrolidone and 0.02% bovine serum albumin (BSA)
- Paraffin oil
- Cellulose nitrate or nitrocellulose membrane filter with uniform porosity. (e.g., Millipore 25 HAWP; nylon membranes used for blotting protocols are available under various trade names from commercial suppliers)
- RNase A (20 pg ml–1 in 2X SSC) for the specific cleavage is needed.
- Restriction enzyme and an appropriate buffer is used.
- Radioactively labelled RNA as a probe for specific detection where autoradiography is done.
Note: Handling of radioactive probes must be careful which necessitates reliable safety measures and legal regulations.
- For detection of RNA labelled with tags such as 3H, 35S, 125I or 14C, there is a requirement of 2,5-Diphenyloxazole (PPO) in toluene at a concentration of 20 % wt/vol.
Equipment
- The transfer from narrow strips of gel can be achieved by using three pieces of glass or Plexiglas having a size of 5 cm × 20 cm with the thickness similar to the thickness of the gel.
- There is a requirement of thick, dry filter paper (four to five in numbers) or paper towels of 10 cm × 18 cm in size.
- Hybridization vessel possess larger dimension (0.8 mm deep × 2 cm high × ~1 cm longer) than that of membrane used for hybridization and the material used for developing hybridization vessel is Perspex (note: Several alternative procedures are followed for hybridization)
- Four narrow pieces of Perspex possessing thickness similar to that of gel. The length of Perspex is sufficient to surround the gel at a spacing of ~3 mm
- A tray having 20–50 mm (approx.) depth and 20 mm (approx.) length and width larger than the gel
- A glass sheet with length sufficient to be placed on the tray and narrower to have a gap of 10 mm on each side
- Several thick pieces of filter paper having a large area as compared to gel. The length of filter paper is adequate to cover the glass sheet and can be dipped within the tray.
- A moistened piece of nitrocellulose membrane, having a wider area which can cover the entire gel. The nitrocellulose membrane is placed on the top of four strips made of Perspex. The moistening of nitrocellulose membrane is done using 2× SSC.
- Paper towels which are stacked one on top of other.
- Apparatus for casting gel.
- Gel tank for carrying out electrophoresis is necessary.
- Power supply for the entire device set up is required.
Reagent Setup
- DNA: The entire procedure is initiated by employing enzyme digested DNA of varying concentrations which will quantify the optimum DNA concentration and specify restriction enzyme to be used. Generally, an amount of 1 µg of DNA derived from clones (e.g. from plasmid or bacteriophage clones) is adequate enough for plasmids having low-copy-number. We require larger amounts for carrying out the separation of complex DNA (e.g. genomic DNA). The advisable range to be considered would be 5–10 µg.
- The electrophoresis buffer used is TAE which has a composition of 40 mM Tris, 20 mM acetic acid, 1 mM EDTA with pH range of 7.4–8.2 which is normally made as stock concentration of 20X or TBE made up of 89 mM Tris, 2.5 mM EDTA, 89 mM borate, normally made as a 10 × stock).
- TAE is recommended to be best when we run gels for a shorter interval of time and when the recovery of DNA fragments from gel is to be carried out.
- TBE is considered to be a better buffer especially when we have to run the gels for a time period exceeding 2 h.
Procedure
Step 1: DNA purification
To extract the DNA present inside the nucleus of a cell, we must first lyse the entire cell to enable the expulsion of the DNA. Incubating the cell culture with detergent lyses the entire cell. Now the lysed sample contains DNA, protein, and debris. Protein is lysed by adding the proteinase enzyme and incubated. DNA is purified and separated by alcohol precipitation and fibers are removed by using a buffer.
Apart from standard manual isolation procedures, there are commercially available kits like GenElute™ for the isolation of DNA from a variety of sources such as mammalian cells, plants, bacteria, fungi. High purity of the isolated DNA is ensured in case of use of commercial kits.
Step 2: Fragmentation
The long nucleotide sequences should be broken into smaller fragments for the purification or identification process. This is done by the restriction endonuclease enzyme.
All the reagent necessary for the digestion process should be kept on ice before setting up a restriction digestion reaction with suitable enzyme and appropriate DNA concentration. The components are added to the multiwell plates or microcentrifuge tubes (PCR tubes) and mixed by aspirating the contents with the help of a pipette slowly to avoid formation of any bubbles. It is to be taken care that the enzyme has to be added at the last step and until then it should be stored at −20 °C. To ensure complete digestion of the DNA a surplus amount of enzyme is supposed to be added as the fragments produced due to partial digestion can cause ambiguity in the results leading to inaccurate analysis of the subsequent blot to be analysed. Nevertheless, the concentration of the enzyme added in total should not surpass one-tenth of the total volume of the digest, because the glycerol content generally found in stock of the enzymes has the capacity to impede digestion process when present at high concentration. Also, inorder to minimize the probable errors due to pipetting master mixes should be prepared accordingly when there is a requirement of analysis of a large number of samples.
The incubation of the digests is carried out at 37 °C either in an incubator or a water bath. A water bath is generally preferred. For DNA samples obtained from cloning 1–2 h deemed sufficient. In case of genomic DNA there is a requirement of overnight digestion wherein there is a chance that the enzyme is over in between. To ascertain that the overnight reaction is successful half of the enzyme is to be added at the beginning of the digestion reaction and the second half can be added in the morning and the incubation can be continued for an hour.
Once the digestion is over it may be the case where a concentration step is required to ensure that suitable volume of DNA is present for loading into the gel which is generally fixed at 20 µl per well. This makes the total volume to be in 24 µl after the addition of 6X loading buffer. A standard method for concentrating the DNA samples involve precipitation in presence of ethanol after which the DNA sample is resuspended in ddH2O. The traces of ethanol should be removed completely which otherwise would lead to spilling of the samples out of wells once the samples are loaded.
Step 3: Gel Electrophoresis
Nucleic acids are negatively charged molecules. So they move towards the anode in an electrophoresis chamber. The movement of the DNA fragments differentiates the rate of the transport thus enabling the separation by size.
The percentage of the gel that is to be used and size of the gel has to be determined. The percentage will decide on the size of the fragments that will be separated and the size of the gel on the other hand will give the range of fragments that are feasible to be resolved. Longer gels are generally needed in the case of separation of genomic DNA or multiple fragments that are having similar sizes inorder guarantee appropriate separation. Generally, a 0.7–2% gel is considered to be adequate for most of the applications. However in case of some genomic DNA samples it may be required that a low percentage of gel is supposed to be run for the necessary separation of the fragments. When a less than 0.8% gel is required to be run a high base gel percentage is required (upto 2%) to provide support as low percentage gels are very fragile. It is necessary that the base gel is dispensed before the comb is positioned. As soon as the base gel is set the low percentage gel is poured on top and the comb is placed in a way that it does not touch the base gel at the bottom.
- A 1X electrophoresis buffer is made after dilution of the prepared stock solution in ddH2O and agarose is added to it in a conical flask which is kept under constant stirring to avoid formation of clumps. Thereafter, the agarose is melted in a microwave or on a stirrer with heating under constant stirring. In case a microwave is used the gel should be swirled after every 30s to ensure even consistency is maintained. In most of the standard microwave the gel completely melts within 3 min.
Note : the agarose should be checked regularly during heating as it is susceptible to boiling in a short time. To ensure there is no spillage it is recommended that a conical flask sufficiently larger in volume is to be used during agarose preparation.
- The flask containing the molten gel is required to be transferred to a magnetic stirrer and the gel is allowed to cool down slowly to around 60ºC under gentle but constant stirring. The ethidium bromide is supposed to be added to the gel at this stage, at a concentration of 5 µg ml–1.
- By the time the gel is cooled down the 1X electrophoresis buffer is prepared in an appropriate amount to fill the tank reservoir upto the level of few millimetres above the gel slab to ensure proper immersion,
- The gel casting tray is prepared with a comb of suitable teeth size depending on the sample volume to be loaded. The casting tray should be in a straight platform before pouring the gel onto it. The formation of bubbles is supposed to be avoided as far as possible as they render the DNA to run in an irregular manner. The bubbles in case formed, can be by burst by using a pipette tip and this is to be done as soon as the gel has been poured.
- As soon as the gel is set it can be understood by observing its opalescent appearance and the comb should be gently removed. The gel is then transferred to the tank and 1X running buffer is added to it inorder to immerse the gel. The buffer should be added in sufficient amount keeping in mind the maximum capacity of the tank because it increases the buffering effect as well as protects the gel from melting down due to generation of heat particularly if the gel is to be run for longer period of time. One has to be careful while removing the combs so as to not tear the wells. In case the gel apparatus in use provides for removing the comb after the gel is placed in the tank and immersed in the buffer the comb should be removed in the final step to avoid breakage of the wells . When a gel of lower percentage is used, before removal of comb a brief refrigeration could help in sustaining the wells in proper shape.
- The DNA samples are prepared by adding 1X loading buffer for each 5 µl of sample. The loading dye and the sample should be mixed well without formation of any bubbles which will eventually lead to spilling of the samples from the wells once they are loaded.
- The samples are then loaded carefully into the wells and one or preferably two of the wells are left blank for the molecular weight marker to be loaded which generally comes in a ready to load formulation. While loading the pipette tip is supposed to be placed at below the corner at the top of the well so that the pipette tip does not pressurise against the bottom of the well which may also lead to perforation of the well. The sample is to be loaded slowly to avoid spillage out of the wells and mixing of samples with the contents of the next well.
- The lid is then placed on top and connected to the power pack with the voltage set at an appropriate level. Inorder to ascertain if the setup is working fine small bubbles can be seen popping up as the power supply is switched on. In case of genomic DNA, the voltage should be set at low preferably not exceeding 20 V and for plasmid DNA it can be around 100V. The electrodes should be located in correct position for the DNA to run in the appropriate direction. The positive socket is generally red and the negatively charged one is black in color.
Step 4: Denaturation
DNA thus attained are double stranded in nature. For our purpose of probe hybridization, we need a single stranded DNA. DNA is therefore denatured in an alkaline solution. This results in the formation of denatured DNA.
Step 5: Blotting
Blotting is the transfer of the fragmented DNA sequence to the nitrocellulose membrane or nylon membrane. The process is done by either electroblotting or capillary blotting. The DNA molecule is saturated using a NaCl solution and permanently fixed using either UV radiation or drying.
Choice of the membrane for blotting
In the original protocol nitrocellulose membrane have been used for the blotting in case of Southern blot but in recent times nylon membranes have been implemented for the blotting process due to their ability to bind more amount of DNA efficiently which allows the Southern blot to be carried out with less amount of target DNA.
- The preparation time for transfer takes approximately 2–3 h
- In case the gel is run without ethidium bromide, it needs to be immersed electrophoresis buffer mixed ethidium bromide for 0.5–2 h.
- Thereafter, the gel is transferred carefully to a gel documentation system for capturing the photograph of the gel under ultraviolet light of 256 nm although when DNA is present in high amounts it can also be detected at 310 or 365 nm. If a scale is placed along with the gel it helps in co-relating the fluorescence photograph with the final autoradiograph that will be generated.
- If sufficient separation of the DNA has not occurred then the gel can be run for a longer time after returning to the tank. Distinct bands generally appear in case of clonal DNA and genomic
- DNA mostly yields a smear in which the repetitive elements of the DNA are represented by the appearance of brighter bands. Thereafter the portion of the gel to be subjected to blotting can be cut apart using a blade. In the experiments where large fragments needs to be transferred the gel should be subjected to a dilute acid bath for approximately 10 min inorder to bring in depurination.
- In the next step the gel is denatured completely by submerging it for 15–30 min in the presence of 1.5 M NaCl, 0.5 M NaOH in a tray that is placed in a rocking platform.
- The above mentioned solution is then substituted with 0.5 M Tris HCl (pH 7) and 3 M NaCl. It was further incubated for 15-30 min which leads to neutralization of the gel.
The time required for transfer is about 15 min.
- In case of the transfer of strips the constituents of gel transfer are prepared as such that there should be a portion of thick filter paper of appropriate dimension preferably 20 cm × 18 cm dipped in 20X SSC buffer along with a nitrocellulose filter which is equal to the length of the strip of the gel having a width of 1 cm dipped in 2X SSC. The filter paper is required to be immersed and the membrane should be transferred by initially floating them on the solution surface to avoid uneven transfer due to the trapped air.
- The large filter paper is laid on a glass or plastic platform after being soaked in 20× SSC in case of narrow strips. Utmost care should be taken so that no air is trapped below. In case of wider strips, the tray is first filled with 20× SSC before a glass plate is laid on to it. Thereafter, a compact arrangement of filter paper soaked in 20× SSC is placed on it before being dipped into the tray.
- A glass or a Perspex sheet is placed on uppermost and towards one of the sides of the wet paper.
- The gel is then removed from the above neutralizing solution and is laid parallel to the glass approximately 2–3 mm apart. Similarly, on the opposite side of the gel, the second glass is laid around 2–3 mm apart.
- The nitrocellulose membrane is then laid on the top portion of the gel with the ends balanced on the glass forming a between the air space. Air should not be trapped between the membrane and the gel hence the alignment has to be carried out with care. The membranes should not be shaken once it is placed.
- The absorbent paper is finally placed on the top. It is to be noted that placing a heavy weight on the top may disorient the gel in turn affecting the final results.
- The transfer process is then started for approximately 3 h which vary depending on the concentration of the gel as well as the size of the fragment in question. Inorder to avoid drying during the transfer 20× SSC must be replenished to avoid shrinkage of the gel. During this process of the addition of buffer, it must not fill the gap between the gel and the glass or sheets.
- Once the transfer is completed the nitrocellulose membrane is removed carefully in a way that the gel should remain attached.
- Then the nitrocellulose membrane is turned and the boundaries of the gel is traced using a pencil
- The gel is then removed from the nitrocellulose membrane and if some amount of DNA is left out during the transfer it can be visualised under UV light as explained before.
- The portion of the nitrocellulose membrane which was touching the gel should be removed using a blade.
- The cut portion is then kept immersed in 2X SSC for approximately 10–20 min before it is subjected to baking at 80 °C for 2 h in a vacuum oven. In another way the DNA can also be fixed via UV crosslinking mediated by exposure to short wavelength UV light in a commercial set up. If desired the dry membranes can also be stored at room temperature for future reference
Once the transfer process is completed the preparation for hybridisation reaction is to be carried out. For this a blocking step that usually takes around an hour is mandatory for eliminating non-specific reactions. This step is also known as the prehybridization step.
The membrane is incubated in standard Denhardt’s solution for 1 h or for certain cases more time is required depending on the type of reaction. Salmon sperm DNA is also widely applied as a blocking agent. Commercially available prehybridization solution like PerfectHyb™ Plus buffer is used for preparing blocking solution containing Salmon sperm DNA. Briefly after incubating the prehybridization solution to 42°C the heat snap chilled salmon sperm DNA is added to it at a concentration of 50 µg/mL. The prehybridization solution containing the sperm DNA is allowed to interact with the blotted membrane inside a hybridization chamber for upto 5 h.
Step 6: Hybridization
Labeled probe is added to the membrane buffer and incubated for as It takes several hours for the probe to find the exact target sequence.
The time required for hybridization usually 1–16 h depends on factors like complexity of the probe and concentration
The radioactive RNAs are present in limited amount as it is derived from cells making it an important factor for the original protocol. Due to this the volume of the hybridisation solution used is supposed to be less so that the desired RNA concentration can be achieved. There can be two approaches for hybridisation experiment where one uses smaller volume and the other utilizes a vessel destined to hold the membrane in low volume of liquid. The suitable set up possess a plastic bag that can be sealed using heat and it consists of optimum amount of hybridisation mix and can be dipped in a water bath or a hybridisation tube with capped ends. In case of the tube it must be ensured that the membrane is wet when the tube is subjected to rotation cycles in a rotisserie oven. The probes applied can be either RNA or DNA. The cells act as a source for the RNA or alternatively, they can also be obtained from clones or PCR products via in vitro transcription In case of DNA they can be either clones or PCR products. The procedure adopted for the labeling method relies on the on the source from which the probe is derived.
- In case of hybridization by immersion in paraffin oil a drop of solution containing the probe RNA of appropriate concentration depending on the membrane size is placed on a plastic sheet.
- One of the ends of the nitrocellulose membrane is allowed to soak the liquid from the drop by slowly moving the membrane over the drop surface. Once it is wet the same is repeated for the other side of the membrane.
- The membrane is then dipped in hybridization solution saturated with paraffin oil at a temperature of 40–65 °C which depends on the type of the solvent in use. Here it is to be taken into account that when the membrane is baked in presence of 2X SSC that leads to the introduction of salt which in turn will be the deciding factor for the solvent in which RNA will be dissolved. This process yields good results but suffers from disadvantages like uneven background which can be solved if the hybridization is processed at 40 °C in presence of 2X SSC and 40% formamide. This method also works well in case of hybridization involving large fragments.
- In case of hybridization using a vessel destined to place the membrane in presence of a small quantity of liquid.
- The vessel is initially filled with the solvent that is required for hybridization to take place.
- The membrane is then fed into it through a thin aperture at the top.
- The solvent is then drained off and the probe solution is added. The amount of the solution needed depends on the size of the membrane. For wider membranes this type of vessel is not suitable instead they can be hybridised inside a close-fitted cylindrical tube for transferring several gels are too wide to be hybridized in this type of vessel. In case the hybridization is carried out inside a water bath the top is sealed as the water bath contains a lid. Moreover another advantage is that in this case RNA can be reused.
- The membrane is then subjected to hybridisation for a suitable period of time depending on the concentration and purity of RNA as well as other conditions. It is appropriate to leave it overnight for most of the cases.
- Once the hybridisation period is over the membranes are removed and the blotted against filter papers
- Thereafter, it is washed for 20–30 min using large amount of solvent used in hybridisation and at the same temperature at which hybridisation is carried out.
- The excessive amount of the probe is washed using the sodium chloride and buffers comprising of detergents. In case the salt concentration is lowered the wash is considered to be more stringent and more amount of such nonspecific probe that is bound to the membrane is removed with a gradual increase of the number of stringent washes. Application of a high temperature while washing also adds up to the stringency of the wash. Once the wash is completed only the specific hybrids remain on the blot and the non-specific ones are removed.
- The membrane is checked for background using a radiation monitor. In case it is high the membrane is treated with RNase for 30 min at 20 °C and then rinsed using 2X SSC.
- Finally the membranes are air dried.
Detection time is approximately 1–48 h, which mostly depends on the nature of the probe
- In detection of radioactivity of RNA there are two options namely 32P-labeled RNA detection and 3H-, 35S-, 125I- or 14Clabeled RNA detection.
- For the detection of 32P-labeled RNA the membrane is wrapped in plastic with no air bubbles trapped in it and laid on X-ray film. With the application of slight pressure the membranes are flattened.
- For Detection of 3H-, 35S-, 125I- or 14C-labeled RNA fluorography is implemented. The membranes are soaked in a solution of PPO in toluene and then the membranes are air dried. Thereafter, they are laid on the X-ray film and maintained at –70 °C
The approximate timings for all the processes are crucial in obtaining good results.
- The restriction digestion usually takes 2–24 h which can comprise of overnight digestion.
- Similarly in case of electrophoresis the timing can vary between 1–16 h and can be implemented overnight
- The preparation for the transfer takes around 0.5–3 h
- For setting up the transfer apparatus approximately 15 min is required.
- Transfer time can be between 3–16 h including overnight transfer.
- Blocking to reduce non-specificity can be done for upto 1 h
- The hybridisation process takes 1–16 h and as per the probe nature and amount it can continue overnight.
- The detection takes around 1–48 h which is affected by the yield in hybridisation and probe specificity.
Troubleshooting
For troubleshooting of the process the following steps can be considered
- The samples may leak out of the wells due to damage or puncture of the wells. One has to be careful while removing the comb as well as while loading the gel.
- Floating of the samples out of the well is commonly observed due to residual ethanol and also due to loading of the gel in haste. The ethanol is to be completely removed before sample loading and the gel should be loaded slowly.
- The samples are not run straight and bands have frowned appearance. This may happen when the gel is run at too high voltage and the problem can be solved by choosing a lower voltage.
- DNA including the molecular weight markers are not visible. This may be due to less amount of ethidium bromide used which can be resolved by use of appropriate concentration.
- In case where the molecular weight markers can be seen but the samples cannot be visualised it may be that the DNA is degraded. The sample quality of the DNA must be analysed.
- If the DNA as well as the molecular weight markers are not separated, it may be due to the fact that the gel is made up of water instead of 1× running buffer.
- The sample DNA is not separated but the markers are separated. This might be the case when the sample DNA concentration is too high which cause the DNA to stick to the wells.
- In case of an appearance of a background with spots it might be due to the powder from gloves hence gloves without powder is recommended. Increasing the number of washes can also eliminate the background.
- The signal intensity is less. This might be due to less sample content and hence DNA concentration should be increased.
- Also a shorter transfer time or hybridisation timing can affect the signal intensity. Hence, transfer should be be allowed for a longer period as well as should be allowed to proceed for appropriate time. The hybridisation temperature can also be optimised for better signal.
Reuse of Blots
In case of analysis of clinical samples where there is less amount of target DNA it might be advisable to re-use the blots. For this purpose the probe can be removed by warm alkali treatment for a brief interval of time. Thereafter, the membrane undergoes a neutralisation step and then it can be hybridised with a different probe.
Comparison of Southern Blotting with PCR and Chromatography
In most of the applications PCR technology has replaced the Southern Blotting technique. However, PCR technology is dependent on the accurate DNA sequence information for design of specific primers but Southern blotting is independent of such a requirement. Also, in PCR if the design of the primers fail considering the gene rearrangement that is being analysed, Southern blot will be able to provide at least some information. In comparison to chromatography the advantage of Southern blot method is that it amplifies the detection limit levels that are necessary or useful for investigating particular genomic loci. But this method faces drawbacks such as requirement of high starting concentration of target DNA and the method is also dependent on the efficiency of the restriction enzyme in use which in turns effects the results.
Results
The hybridization of probe to the specific DNA on the membrane results to a conclusion that the probe is specific to a fragment on the membrane due to the presence of a complementary sequence in the fragment . A probe that has hybridised with a single fragment of DNA not being digested by restriction enzymes will result in only one band in the final blot. In case the probe binds to many of the similar sequences it will result into multiple bands. The probe specificity as well as decreased non-specific hybridisation is generally modulated by optimising the temperature at which the hybridisation is carried out as well as the salt concentration also needs to be regulated.
Below is an example of a typical Southern blot result.
Herein, human DNA hypervariable fingerprints were digested with Hinf I and/or Sau3A and thereafter the Southern blot was hybridized with single stranded DNA probes labelled with 32P. Here, each represents a human minisatellite containing tandem repeats of closely connected variants of a consensus sequence.
Application
Southern blotting is used in a number of applications. The primary usage of Southern blotting is to identify a specific DNA in a DNA sample. It is mostly used in the identification of viral infection and certain bacterial infections. In rDNA technology, The Southern blotting technique is used to isolate a particular DNA. It is also useful in the study of mutation and gene rearrangement, this property is used to diagnose neonatal disease and genetic disease. Due to the precision in DNA identification this technique is used in phylogenetic studies, paternity & maternity analysis, forensic studies and personal identification.
Southern blotting can be applied in studying structure of a gene or to elucidate restriction enzyme maps. Particularly, Southern blotting can be used in the identification of the methylated sites present in case of some genes in particular. Applying restriction nucleases such as MspI and HpaII, which can both identify and cleave among the identical sequence this can be implemented. The discovery of RFLP s by southern blotting has helped in the mapping of several genomes that were crucial to be mapped. In the arena of immunology the clonal rearrangements of the immunoglobulins as well as the T Cell receptor genes that play an important role in eliciting an immune response can be analysed by Southern blotting.