Introduction
The term ‘plasmid’ was coined by Joshua Lederberg in 1952. Originally evolved from bacteria, plasmids are extrachromosomal genetic elements present in most species of Archae, Eukarya and Eubacteria that can replicate independently. Plasmids are circular double stranded DNA molecule that are distinct from the cells chromosomal DNA.
The structure and function of a bacterial cell is directed by the genetic material contained within the chromosomal DNA. In some cases plasmids are generally not essential for the survival of the host bacterium. Although not essential, plasmids contribute significantly to bacterial genetic diversity and plasticity by encoding functions that might not be specified by the bacterial chromosomal DNA. Plasmids specify traits that allow the host to persist in environments that would otherwise be either lethal or restrictive for growth. For example antibiotic resistance and protein expression. Antibiotic resistance genes are often encoded by the plasmid, which allows the bacteria to persist in an antibiotic containing environment, thereby providing the bacterium with a competitive advantage over antibiotic-sensitive species. As a tool, plasmids can be modified to express the protein of interest (e.g., production of human insulin using recombinant DNA technology).
Plasmids have served as invaluable model systems for the study of processes such as DNA replication, segregation, conjugation, and evolution. Plasmids have been pivotal to modern recombinant DNA technology as a tool in gene-cloning and as a vehicle for gene–expression.
Characteristics of Plasmid
Plasmids present in the bacterium differ in their physical properties such as in size (kbp), geometry and copy number.
Plasmid Size
Plasmids range in size from 1 kbp (kilo base pair) to 1000 (kilo base pair) megaplasmids that are many hundred base pairs in size.
Plasmid Geometry
Although most plasmids possess a circular geometry, there are now many examples of plasmids that are linear in a variety of bacteria. Plasmid DNA may appear in one of the five conformations nicked open circular DNA which has one strand cut, relaxed circular DNA is fully intact with both strands uncut, but has been enzymatically relaxed, linear DNA has free ends, supercoiled DNA is fully intact with both strands uncut, and supercoiled denatured DNA is like super coiled DNA, but has unpaired regions that make it slightly less compact.
Plasmid Copy Numbers
Copy number refers to the average or expected number of copies per host cell. Plasmids are either low, medium or high copy number. Knowing which category plasmid falls under is very important when starting out an experiment. If working with a low-copy number plasmid which is associated with a low yield and might therefore be required to set up more cultures. On the other hand, if a poor yield is obtained from a high copy plasmid, troubleshooting is required. In bacterium with high copy number plasmids, during cell division the plasmids get segregate randomly in the daughter cells, whereas bacterium with low copy numbers, during cell division and partition the plasmids divided equally in the daughter cells. An advantage of high copy number is the greater stability of the plasmid when random partitioning (i.e. partitioning of plasmids into daughter cells) occurs at cell division.
Plasmid Isolation
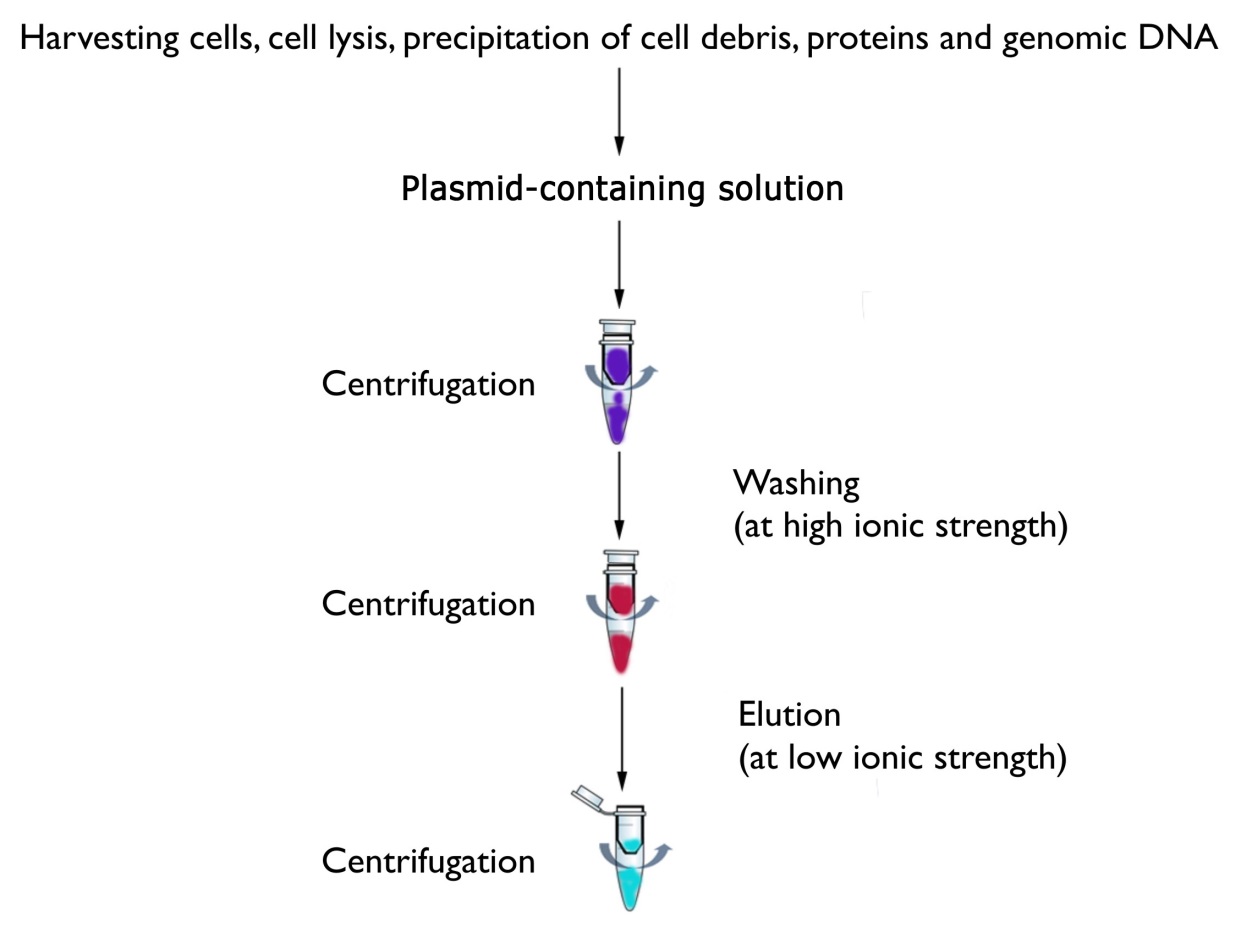
The isolation of plasmid DNA from bacteria is a crucial technique in molecular biology and is an essential step in many procedures such as cloning, DNA sequencing, transfection, and gene therapy. These manipulations require the isolation of high purity plasmid DNA. The purified plasmid DNA can be used for immediate use in all molecular biology procedures such as digestion with restriction enzymes, cloning, PCR, transfection, in vitro translation, blotting and sequencing.
Alkaline lysis is a method used in molecular biology, to isolate plasmid DNA or other cell components such as proteins by breaking the cells open. Bacteria containing the plasmid of interest is first grown, and then allowed to lyse with an alkaline lysis buffer consisting of a detergent sodium dodecyl sulfate (SDS) and a strong base sodium hydroxide. The detergent cleaves the phospholipid bilayer of membrane and the alkali denatures the proteins which are involved in maintaining the structure of the cell membrane. Through a series of steps involving agitation, precipitation, centrifugation, and the removal of supernatant, cellular debris is removed and the plasmid is isolated and purified.
Principle
Purification of plasmid DNA from bacterial DNA using is based on the differential denaturation of chromosomal and plasmid DNA using alkaline lysis in order to separate the two. The basic steps of plamid isolation are disruption of the cellular structure to create a lysate, separation of the plasmid from the chromosomal DNA, cell debris and other insoluble material. Bacteria are lysed with a lysis buffer solution containing sodium dodecyl sulfate (SDS) and sodium hydroxide. During this step disruption of most cells is done, chromosomal as well as plasmid DNA are denatured and the resulting lysate is cleared by centrifugation, filtration or magnetic clearing. Subsequent neutralization with potassium acetate allows only the covalently closed plasmid DNA to reanneal and to stay solubilized. Most of the chromosomal DNA and proteins precipitate in a complex formed with potassium and SDS, which is removed by centrifugation.
The bacteria is resuspended in a resuspension buffer (50mM Tris-Cl, 10 mM EDTA, 100 µg/ ml RNase A, pH 8.0) and then treated by 1% SDS (w/v) / alkaline lysis buffer (200mM NaOH) to liberate the plasmid DNA from the E. coli host cells. Neutralization buffer (3.0 M potassium acetate, pH 5.0) neutralizes the resulting lysate and creates appropriate conditions for binding of plasmid DNA to the silica membrane column. Precipitated protein, genomic DNA, and cell debris are then pelleted by a centrifugation step and the supernatant is loaded onto a column. Contamination like salts, metabolites, and soluble macromolecular cellular components are removed by simple washing with ethanolic wash buffer (1.0 M NaCl, 50mM MOPS, pH 7.0, isopropanol (v/v) 15 %). Pure plasmid DNA is finally eluted under low ionic strength conditions with slightly alkaline buffer (5 mM Tris / HCl, pH 8.5).
Culture Media
Yield and quality of plasmid DNA highly depend on the type of culture media used. Most plasmid purification are optimized with cultures grown in standard Luria Bertani (LB) medium. For LB medium preparation dissolve 10 g tryptone, 5 g yeast extract, and 10 g NaCl in 800 ml distilled water. Adjust the pH to 7.0 with 1 N NaOH. Adjust the volume to 1 liter by adding distilled water and sterilize by autoclaving. The cell culture should be incubated at 37 °C with constant shaking (200–250 rpm) preferably 12–16 h overnight. Usually an OD of 3–6 can be achieved. Alternatively, rich media like 2 x YT (Yeast / Tryptone), TB (Terrific Broth), or CircleGrow can be used. Apk
Also cares needs to taken, as overgrowing a culture might lead to a higher percentage of dead or starving cells and the resulting plasmid DNA might be partially degraded or contaminated with chromosomal DNA. To find the optimal culture conditions, the culture medium and incubation times have to be optimized for each host strain / plasmid construct combination individually.
Lysate & Neutralization
Lysis formulas may vary depending on whether you want to extract DNA/RNA/Plasmid. All methods of lysing bacteria will yield plasmid solutions contaminated with chromosomal DNA and RNA. Centrifugation removes the vast majority of chromosomal DNA (it will form a pellet, while plasmid DNA remains soluble), and treatment with RNase will eliminate contaminating RNA.
Generally speaking, lysis buffers contain a high concentration of chaotropic salts. Chaotropes have two important roles in nucleic acid extraction. Firstly, they destabilize hydrogen bonds, van der Waals forces and hydrophobic interactions, leading to destabilization of proteins, including nucleases. Secondly, they disrupt the association of nucleic acids with water, thereby providing optimal conditions for their transfer to silica.
Separation and removal of the plasmids from the bacterial cell is brought about by resuspension of 1-5 mL of culture in a resuspension buffer (50mM Tris-Cl, 10 mM EDTA, 100 µg/ ml RNase A, pH 8.0) and pellet cells in a microcentrifuge at 11000 x g for 30 s.
Lysate is achieved by adding 250 µL of lysis buffer with neutralization buffer, as it aids in complete precipitation of SDS, protein, and genomic DNA. Incomplete neutralization leads to reduced yield. However, released plasmid DNA is very vulnerable at this point and shaking too much or too strongly will damage the DNA.
Binding and Washing in Silica Membrane
After centrifuging the lysate through silica membrane, the desired nucleic acids should be bound to the column and impurities such as protein and polysaccharides should be in the flow-through. While, plant samples will likely contain polysaccharides and pigments, while for blood samples, the membrane may be slightly brown or yellow in color. The wash steps will remove such impurities. There are typically two wash steps, although it varies depending on sample type. The first wash will often include a low concentration of chaotropic salts to remove residual proteins and pigments. This is always followed with an ethanol wash to remove the salts.
Columns contain a silica resin that selectively binds to DNA/RNA. The DNA of interest can be isolated by virtue of its ability to bind silica in the presence of high concentrations of chaotropic salts. These salts are then removed with an alcohol based wash and the DNA is eluted using a low-ionic-strength solution such as TE buffer or water. The binding of DNA to silica seems to be driven by dehydration and hydrogen bond formation, which competes against weak electrostatic repulsion. Hence, a high concentration of salt will help drive DNA adsorption onto silica, and a low concentration will release the DNA.
Elution
The elution buffer volume and method can be adapted to the subsequent downstream application to achieve higher yield and / or concentration that the standard method. Elution buffer is used to wash away unbound proteins at first and at a greater concentration it releases the desired protein from the ligand. It is important that the elution buffer works quickly without changing the function or activity of the desired protein. For maximal DNA elution, allow the buffer to stand in the membrane for a few minutes before centrifugation. Elution Buffer AE (5 mM Tris/HCl, pH 8.5) can be replaced by TE buffer or water as well. Using a weakly buffered slightly alkaline buffer containing no EDTA is preferred especially if the plasmid DNA is intended for sequencing reactions.
Analytical gel analysis
Removing and saving aliquots during the purification procedure is recommended.
If the plasmid DNA is of low yield or low in quality, the samples can be analyzed by agarose gel electrophoresis to determine at what stage of the purification procedure the problem occurred.
Procedure
Harvest Bacterial and Resuspended Cells
- Choose a single colony from a freshly streaked selective plate and inoculate a starter culture of 2–5 ml LB medium containing the appropriate selective antibiotic. Incubate for approximately 8 h at 37°C with vigorous shaking (approx. 300 rpm).
- Dilute the starter culture 1/500 to 1/1000 into 3 ml selective LB medium. Grow at 37°C for 12–16 h with vigorous shaking (approx. 300 rpm).
- Harvest the bacterial cells by centrifugation at 6000 x g for 15 min and remove as much of the supernatant as possible. Resuspend the bacterial pellet in 0.1-0.5 ml of resuspension buffer (50mM Tris-Cl, 10 mM EDTA, 100 µg/ ml RNase A, pH 8.0). The bacteria should be resuspended completely by vortexing or pipetting up and down until no cell clumps remain.
Cell Lysis
- Add 0.25 ml of lysis buffer, mix thoroughly by vigorously inverting the sealed tube 4–6 times, and incubate at room temperature (15–25°C) for 5 min. Do not vortex, as this will result in shearing of genomic DNA. The lysate should appear viscous. Do not allow the lysis reaction to proceed for more than 5 min.
Neutralization
- Add 0.3 ml of neutralization buffer, mix immediately and thoroughly by vigorously inverting 4–6 times, and incubate on ice for 5 min. Precipitation is enhanced by using chilled neutralization buffer and incubating on ice. After addition of neutralization buffer, a fluffy white material forms and the lysate becomes less viscous. The precipitated material contains genomic DNA, proteins, cell debris, and KDS. The lysate should be mixed thoroughly to ensure even potassium dodecyl sulphate precipitation. If the mixture still appears viscous, more mixing is required to completely neutralize the solution. A homogeneous colorless suspension indicates that the SDS has been effectively precipitated.
Load Lysate on Column
- Before loading the column, carefully remove the supernatant and then transfer it to a collection tube containing the column and centrifuge at 13,000 rpm for 1 minute.
- Discard the flow-through liquid and remove supernatant containing plasmid DNA promptly. After centrifugation, the supernatants should be clear.
- If the supernatant is not clear, a second, shorter centrifugation should be carried out to avoid applying any suspended or particulate material to the column. Suspended material (which causes the sample to appear turbid) will clog the column and reduce or eliminate flow.
Bind and Wash
- Add 0.7 ml of wash buffer to the column placed in the collection tube and centrifuge for 10 minutes at 13000 rpm for 1 minute. Equilibrate by applying 1 ml equilibration buffer ( 750 mM NaCl, 50 Mm MOPS, ph 7.0, 15 % isopropanol ) and allow the column to empty by gravity flow. Flow of buffer will begin automatically by reduction in surface tension due to the presence of detergent in the equilibration buffer.
- Apply the supernatant from step 6 to the column and allow it to enter the resin by gravity flow.
Plasmid Elution
- Elute DNA with 0.8 ml elution buffer (1.23 M NAcL, 50 mm Tris-Cl, pH 8.5, 15 %v isopropanol) Collect the elute in a 1.5 ml or 2 ml microcentrifuge tube.
- Precipitate DNA by adding 0.7 volumes (0.56 ml per 0.8 ml of elution volume) of room-temperature isopropanol to the eluted DNA. Mix and centrifuge immediately at ≥10,000 rpm for 30 min in a microcentrifuge. Carefully decant the supernatant. All solutions should be at room temperature in order to minimize salt precipitation.
- Wash DNA pellet with 1 ml of 70% ethanol and centrifuge at 10,000 rpm for 10 min.
- Carefully decant the supernatant without disturbing the pellet.
- The 70% ethanol removes precipitated salt and replaces isopropanol with the more volatile ethanol, making the DNA easier to redissolve.
- Air-dry the pellet for 5–10 min, and redissolve the DNA in a suitable volume of buffer (e.g., TE buffer, pH 8.0, or 10mM Tris-Cl, pH 8.5). Redissolve the DNA pellet by rinsing the walls to recover all the DNA.
Determination of yield
To determine the yield, DNA concentration should be determined by both UV spectrophotometry at 260 nm and quantitative analysis on an agarose gel. To quantitate the nucleic acid concentration, dilute the plasmid DNA 1 : 100 or 1 : 50 (depending on the plasmid copy number) in TE buffer and measure the absorbance (optical density) at 260 nm (A260) and 280 nm (A280). Use TE buffer as the blank. This measurement permits the direct calculation of the nucleic acid concentration using the formula
[DNA] (μg/mL) = A260 × Dilution factor × 50
where 50 is the extinction coefficient of DNA. The ratio A260/A280 provides a reasonable estimate of the purity of the preparation.




