Western Blotting
Brief Introduction: Blotting Techniques
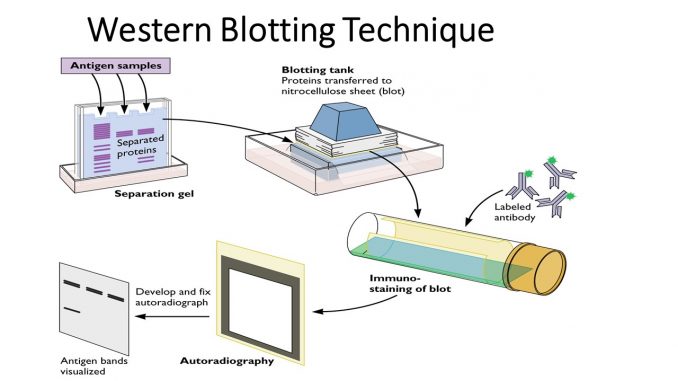
The blotting technique is a tool used in the identification of biomolecules such as DNA, mRNA, and protein during different stages of gene expression. Protein synthesis involves the expression of a DNA segment which gets converted to mRNA to produce the respective protein. Subtypes of blotting such as northern, western & southern depend upon the target molecule that is being sought. When a DNA sequence is a foundation or code for a protein molecule, the particular DNA molecule of interest can be blotted using Southern Blotting technique. During gene expression, when the DNA is expressed as mRNA for a protein production, this process can be identified by Northern blotting. Finally, the coded mRNA produces the concerned protein, this protein identification can be done by Western Blotting.
Western blotting (or immunoblotting) is a widely used method to detect proteins as well as posttranslational modifications on proteins, using antibody based probes to obtain specific information about target proteins from complex samples. It is a routine method in a molecular biology, biochemistry and cell biology field with the multitude of applications. It can provide semi-quantitative or quantitative data about the target protein in simple or complex biological samples.
Since western blotting is a multistep protocol, variations and errors can occur at any step reducing the reliability and reproducibility of this technique. Recent reports suggest that a few key steps, such as the sample preparation method, the amount and source of primary antibody used, as well as the normalization method utilized, are critical for reproducible western blot results. This method relies on the fact that most epitopes (sites recognized by antibodies, generally comprising several amino acids) inspite of denaturation of proteins can still be recognized. Due to high affinities of antibody toward their epitopes, it is a very sensitive method and even picogram quantities of a target protein can be detected. The two primary advantages of western blotting are sensitivity and specificity. Western blotting has advantages over other protein detection techniques. Silver staining, another technique of protein detection detects 10 ng of protein and all proteins in a given sample. Whereas, western blotting can detect as little as 0.1ng of protein, and it selectively detects only the protein of interest. Thus a complex mixture containing only traces of the desired protein may be analyzed accurately with this technique. Western blotting was first described by Harry Towbin in 1979. It was in 1981 when W. Neal Burnette developed an improved version of the method and gave the name “western blotting” simply because of the location of laboratory. Towbin’s landmark paper has been cited more than 54,000 times.
Below is a general procedure for blotting, and every step is critical for obtaining high-quality, reliable and analyzable data.
- Homogenize the sample.
- Separation of the molecule of interest by an electrophoresis membrane.
- Transferring the molecules to a nitro cellulosic membrane/ nylon membrane.
- Hybridization or identification of the molecule
Western blotting is the technique used for separation or identification of protein molecules. This technique can be used for both the active 3D protein and denatured long peptide chains. The 3-D protein in its active structure has sulfur-hydroxyl bonds in the structure. This methodology classifies protein based on the molecular weight and charge. These techniques have application in the identification of a wide variety of infectious diseases like HIV, Hepatitis B, Herpes type 2, feline immunodeficiency disease. The identification of these diseases is done by using the antibody of the particular disease as the probe and these probes are produced in vitro condition. Western blotting is also used for research purpose. This technique can be used in the study of the properties and activity of a protein molecule of interest.
PROCEDURE
Sample Preparation
- Wash cells in the tissue culture flask or dish by adding cold Phosphate Buffered Saline (PBS) and rocking gently. Flask or dish should be kept on ice throughput the process. Discard PBS.
- Add PBS and use a cell scraper to dislodge the cells. Pipette the mixture into microcentrifuge tubes.
- Centrifuge at 1500 RPM for 5 minutes. Discard the supernatant.
- Add 180 ul of ice cold lysis buffer solution to 20 ul of fresh protease inhibitor. This prevents the protease enzyme. Incubate for 30 minutes.
- Incubate for 30 min on ice, and centrifuge this solution for 10 minutes at 12000 RPM at 40c and the sample solution is ready.
- Transfer supernatant (or protein mix) to a fresh tube and store on ice or frozen at -20°C or -80°C.
- Measure the concentration of protein using a spectrophotometer and determine the volume of protein extract to ensure 50 μg in each well.
- Add 5 μL sample buffer to the sample, and make the volume in each lane equalized using double distilled H2O (dd H2O). Mix well.
- Heat the samples with a dry plate for 5 minutes at 100°C.
Gel Preparation
The gel has two parts stacking gel and separation gel. 10% stacking gel and 6% separating gel are generally used. Add the stacking gel solution into the assembly carefully and then add H2O to the top. Wait for 15–30 minutes until the gel solidifies. Overlay the stacking gel with the separating gel, after removing the water. Insert the comb, ensuring that there are no air bubbles. Wait until the gel is solidified.
Electrophoresis
Pour the running buffer into the electrophorator. Place gel inside the electrophorator and connect to a power supply. (Tip: When connecting to the power source always connect red to red, and black to black). Make sure buffer covers the gel completely, and remove the comb carefully. Load marker (6 μL) followed by samples (15 μL) in to each well. Run the gel at 40 volts until the sample reaches the stacking gel and changed into 80 volts from the separation gel. Run the gel for approximately an hour, or until the dye front runs off the bottom of the gel
Fixing and blotting
Fixing is done 5% of bovine serum albumin solution. Cut 6 filter sheets to fit the measurement of the gel and one polyvinylidene fluoride (PDVF) membrane with the same dimensions. Wet the sponge and filter paper in transfer buffer, and wet the PDVF membrane in methanol. Separate glass plates and retrieve the gel. Create a transfer sandwich as follows – Sponge, 3 Filter Papers, Gel PVDF, 3 Filter Papers (Ensure there are no air bubbles between the gel and PVDF membrane, and squeeze out extra liquid). Relocate the sandwich to the transfer apparatus, which should be placed on ice to maintain 4°C. Add transfer buffer to the apparatus, and ensure that the sandwich is covered with the buffer. Place electrodes on top of the sandwich, ensuring that the PVDF membrane is between the gel and a positive electrode. Blotting is carried in two ways, capillary blotting or through electroblotting. Usually, electroblotting is carried out at 40 volts. For capillary blotting, the gel is stacked in the following order: electrophoretic gel followed by blotting membrane followed by wet tissue and lid glass plate.
Blocking and Incubation
Block the membrane with 5% skims milk in TBST for 1 hour. Add primary antibody in 5% bovine serum albumin ( BSA) and incubate overnight in 4°C on a shaker. Wash the membrane with TBST for 5 minutes. Do this 3 times. (Tip: All washing and antibody incubation steps should be done on a shaker at room temperature to ensure even agitation). Add secondary antibody in 5% skim milk in TBST, and incubate for 1 hour. Wash the membrane with TBST for 5 minutes. Do this 3 times.
Detection
Detection and identification can be carried out by a number of methods like radiography, chemiluminescence, colorimetric and x-ray methods.
Reagents Required
- Washing Buffer – Blocking buffer + 0.1% Tween®-20
- Ponceau S – Ponceau S (Sigma P3504), 0.1 g
Acetic Acid, 5 ml
Distilled Water, 95 ml
*Note: Ponceau S is light sensitive.
- PBS – Sodium Chloride, 8.0g
Potassium Chloride, 0.2g
Disodium Potassium Phosphate, 1.15g
Potassium Dihydrogen Phosphate, 0.2g
Distilled Water, 1 liter.
- PBST – Sodium Chloride, 8.0g
Potassium Chloride, 0.2g
Disodium Potassium Phosphate, 1.15g
Potassium Dihydrogen Phosphate, 0.2g
Tween-20®, 1.0ml
Distilled Water, 1 Liter
Principle
The principle of the western blotting is based around a few broad steps: (a) the extraction of cellular proteins from a complex mixture of intracellular and extracellular proteins (from tissue, cells, etc.); (b) quantification of protein concentration and electrophoretic separation of proteins within a gel matrix; (c) transfer to a membrane with a high affinity for proteins; (d) “blocking” the membrane to reduce non‐specific binding; (e) antigen detection by antibodies specific for the protein(s) of interest; (f) incubation with a secondary antibody linked to a label (e.g., chemiluminescent or fluorescent); (g) development and detection of the signal, which is theoretically proportional to the degree of antigen/antibody binding; and (h) quantification of the resulting bands.
Sample Preparation – This is probably the often overlooked step of reproducible western blotting. The method of sample collection depends on the sample type: human tissue, samples dissected post‐mortem and cell culture using scrapers. Once tissue samples are harvested, they should be immediately washed in an ice‐cold neutral pH buffer, before removal of visible fat, snap frozen in liquid N2, and stored at −80 °C. These steps are designed to limit protein degradation and preserve post‐translational modifications (PTMs). In order to obtain robust data, samples should remain frozen until use and undergo as little manipulation as possible (to minimize degradation as indicated by gel streaks). Sometimes, it may be viable for fresh tissue to be utilized for western blotting sample preparation; however, it is more common and preferable to snap freeze tissues. The extraction of proteins of interest from tissue requires the lysis and disruption of cell membranes using homogenization techniques, typically in the form of mechanical, sonication, and/or chemical approaches. Sonication utilizes high frequency sound waves to disrupt cellular membranes, nonetheless, this should follow mechanical homogenization to remove large pieces of tissue that would not be disrupted by sonication alone. Notably, homogenization of muscle cell cultures does not require the same degree of mechanical lysing. Vigorously pipetting the collected cells through a small gauge syringe or fine tip gel‐loading pipette is typically sufficient. Tissues should be homogenized in a buffer designed to solubilize and optimize preservation of the target proteins. Accurately buffering a homogenization solution proximate to the isoelectric point of proteins (the pH at which they have neutral charge), is necessary (pH 7–9) to ensure solubility, and prevention of protein precipitation, through maintenance of positive or negatively charged amino acid functional (R) groups. Addition of non‐ionic detergents (i.e., Triton X‐100) are used to increase solubility of non‐polar insoluble proteins. Proteins retaining tertiary and quaternary structures remain soluble in water, since non‐polar hydrophobic regions are generally oriented toward the center of the protein or within cell membranes. Thus, reducing agents [e.g., dithiothreitol (DTT)] are used to breakdown disulfide bonds (S‐S) between cysteine residues, while sodium dodecyl sulfate (SDS) detergent is added to coat hydrophobic regions of proteins with negative charge and overwhelm positive charges in proteins; this aspect is crucial for resolving proteins in accordance to their molecular mass. Reducing agents are required in subsequent sample preparation stages and may interfere with determining the protein content, and thus it is recommended where possible to avoid or minimize addition before quantification. The reproducibility of western blotting experiments and result interpretation is substantially influenced by an efficient protein extraction and purification step. In order to have an effective proteins extraction, a suitable homogenization method should be selected. The homogenisation method selected should efficiently release the intracellular contents of the cell. Additionally, an optimal lysis buffer should be chosen which can facilitate the proper solubilization of proteins and prevent proteolytic degradation in order to obtain high amounts of target proteins. Protein is extracted from Cell or tissue by mechanical or chemical lysis method. Protein sample is best prepared in fresh lysis buffer in the presence of protease inhibitors. If one is interested in probing phosphorylated proteins phosphatase inhibitors are used. An optimal lysis buffer should efficiently extract the target proteins. In some cases the lysis buffer should possess the ability to maintain the integrity of protein as this is important for doing western blots on protein complexes separated by native gels. Sample can be prepared using either denaturing or native lysis buffer. Denaturing buffer contains SDS in it whereas native buffer lacks it. Lysis buffer can be optimized by varying the concentration of detergents and other chemical components to solubilize target proteins and prevent protein precipitation. The use of sodium chloride (NaCl) or potassium chloride (KCl) at a concentration of 100 mM to 150 mM in homogenization buffers has been reported to prevent protein aggregation. Reducing agents such as dithiothreitol (DTT) are often utilized to help solubilize proteins as well as to keep some proteins in an active state (such as for native gel electrophoresis). These reducing agents often interfere with protein quantification methods utilized. Centrifugation at 16000 x g for 15 min at 4ºC is performed to clear off the lysates. The expression, conformation, and stability of proteins varies significantly with the buffer and experimental conditions utilized. The principal factors that can influence sample processing include the time between tissue collection and its freezing, frequent freeze/thaw cycles of tissue samples, poor homogenization techniques, and use of inappropriate lysis buffer. After the clear lysate is collected protein quantification is performed utilizing different methods including the Bradford assay, UV absorbance (Nanodrop) method, bicinchoninic acid (BCA) method, and ortho-phthaladehyde assay. Appropriate lysis buffer compatible with subsequent chosen protein assays must be used as blanks and in the standard curves to avoid interference by buffer components during protein quantification. The presence of insoluble and soluble proteins in samples will also affect protein quantification. The concentration of protein is determined by spectroscopy. Commonly used quantification methods are Bradford and BCA. Bradford is simpler and faster procedure and more commonly used whereas, BCA is less variable and less susceptible to detergents. After protein quantification sample is prepared by adding 5X sample buffer used for SDS-PAGE to achieve final concentration of 1X. Loading buffer contains glycerol which helps to sink the sample in well. Tracking dye (bromothymol blue) is also added in sample to monitor the movement of proteins. One imp aspect of sample preparation is the final protein concentration of the samples. The sample concentration of 1 to 1.25 mg/ml is good in order to load 10 -20 ul per well. Typically 10-50 ug of total lysate protein per well should be loaded.. Samples must be normalized to have the same concentration so that the loading vol is consistent across the samples in the gel. If loading vol changes between samples it will affect the running of each sample that has allow vol. Protein in sample buffer can be stored at -20ºC.
Gel electrophoresis – It is the first part of the western blotting technique. It can be 1D or 2D, in 1D protein molecules move in the singular axis based on both molecular weight and charge. Whereas 2D begins with electrophoresis in the first dimension and then separates the molecules perpendicularly from the first to create an electropherogram in the second dimension. In the first dimension at isoelectric point, the protein molecule acquires a certain charge based on pH. At this point when a constant voltage is applied the protein molecules move towards the electrode. This property leads to the separation of the protein molecule. The size of the protein molecule hinders the rate of movement. So molecular weight also acts as a factor for separation rate in electrophoresis. In the second dimension, the molecules are then separated at 90 degrees from the first electropherogram according to molecular mass. Since it is unlikely that two molecules will be similar in two distinct properties, molecules are more effectively separated in 2-D electrophoresis than in 1-D electrophoresis. The extracted sample in the sample preparation stage is loaded in the well of SDS-PAGE (Sodium dodecyl sulphate- polyacrylamide gel electrophoresis) which allows resolution of denatured proteins by their molecular weight using an electric field and a porous acrylamide based matrix. The protein extracts are mixed with SDS, an anionic detergent that binds uniformly to proteins approximately 1 SDS molecule for every 2 amino acid residues. SDS has two roles – disruption of tertiary structure of protein in to linear molecules and coating of protein with a negative charge. Further protein is unfolded by reducing disulphide linkages present in protein with the help of thiol -SH group-containing compounds such as beta-mercaptoethanol or DTT. Finally samples are heated to 75-90ºC to maximize denaturation and enhance uniformity of coating by SDS. The mobility of each protein depends on the net charge of the molecule, the voltage of the field and the resistance of the solution in which it is immersed. Negatively charged SDS treated protein migrate through PAGE toward the positive anode. Protein with less molecular weight migrates faster than proteins with higher molecular weight. PAGE is inert, thermostable and transparent which makes it an ideal choice for gel matrix. It can produce a wide range of pore sizes which allows for separation and resolution of the protein of interest. The pore size for PAGE is determined by the amount of total acrylamide and amt of crosslinker bis-acrylamide present in the gel. A higher percentage of total acrylamide has smaller pores and is used for resolution of smaller proteins. Most commonly used acrylamide to bis-acrylamide ratio for SDS-PAGE is 37.5:1, whereas the 19:1 gel used for denaturing DNA/RNA gels and 29:1 gels are used for native DNA/RNA gels. Whereas electrophoresis of nucleic acids utilizes a constant pH within the buffer and gel to achieve separation protein samples require a discontinuous buffer system. Discontinuous systems utilize gels separated into two regions, comprising a “stacking gel” above a “resolving or separating gel” with larger and smaller pores, respectively. Discontinuous systems are designed to focus protein samples and allow clear resolution of proteins. Stacking and resolving gels differ in terms of gel percentage and pH. Stacking gel has a 4-5% acrylamide and a pH of 6.8 whereas resolving gel has higher % acrylamide in range of 6-15% and a pH of 8.8. Stacking gels minimize charge on protein thereby allowing them to “stack” on top of each other, resulting in sharper bands. Gels can be commercial precast or hand cast. Commercially available gels i.e. precast gels come in different sizes, percentage and formulations. Gradient gels, which are available commercially detect large and small proteins in the same sample on the same blot. They are good choice for probing multiple protein with major differences in their molecular weight. Some commercially available precast gels don’t use stacking and they have a gradually increasing acrylamide increasing percentage (4-20%) from top to bottom giving it a gradient nature. Gel percentage selection is a key factor in achieving optimal separation of target protein. It depends on size of protein of interest.. Handcast gels are low cost and can be readily customized based on requirement. Gels are poured between two glass plates and a comb is placed on the top to create wells for loading samples. The thickness of gel along with number and size of wells determine number of samples and volume that can be loaded onto the gels. In a handcast gel usually gel runs at lower voltage 70-80 V at the beginning so that proteins enter the stacking gel with minimal distortion of the bands. For the commonly used mini gels resolving gels are run at 100 V. As a rule of thumb 10-15 V per cm of gel should be used for resolving gels. For high concentration or high viscosity samples gels should run slower in order to avoid streaking of bands. To run a gel combs should be carefully removed without damaging wells. Any semi polymerized gel residue should be recovered by either rinsing wells or aspiration. Molecular weight markers are very important component of a gel electrophoresis. They are mixture of highly purified proteins which resolves as ladder of bands between 10-250 kDa. Two types of available markers are pre-stained and unstained. Unstained markers are cheaper in comparison to pre-stained markers. Pre-stained markers give advantage to monitor protein resolution in gel in real time. They determine transfer efficiency without the need to stain with Ponceau S. Unlike pre-stained markers unstained markers are not be visualized in real time and need to be stained using Ponceau S. In comparison to pre-stained markers unstained markers give more precise molecular weight estimation.
Transfer – Once the electrophoresis is done the next step is blotting. Blotting is carried out by two mechanisms electroblotting and capillary blotting. Electroblotting uses electric energy to pull out the protein molecule from the gel to the blotting membrane. Capillary blotting uses passive transport of protein molecules together with water molecules as water high concentration moves to lower concentration. Capillary blotting takes a lot of time so this technique is rarely used these days. The membrane is placed directly upon the gel ensuring a mirror image transfer of proteins occurs. Proteins need to be transferred to increase the robustness of the protein-carrying matrix, simplifying the handling process and provide greater epitope accessibility to the antibodies. The separated molecules are transferred or blotted onto a second matrix, generally a nitrocellulose or polyvinylidene difluoride (PVDF) membrane as they have high binding capacity for proteins. They provide better mechanical support and allow the blot to be re-probed and stored. Washing becomes very important in case of PVDF membranes because it gives higher background. Nitrocellulose is used for its high affinity for protein and its retention abilities. However, it is brittle, and does not allow the membrane to be used for re-probing. PVDF membranes bind proteins through hydrophobic interactions and are capable of binding greater amounts of protein (~150 μg/cm2) than other substrates. Although the use of PVDF membranes may be advantageous in capturing greater amounts of protein, membrane–antibody interactions are more likely to occur, generating higher backgrounds when exposing the blots, consequently increasing the importance of performing thorough wash steps. The hydrophobic nature of PVDF membranes requires an initial pre‐soaking in methanol to allow the infiltration of the buffer and the binding of proteins. Another variable to consider is the choice of pore size (i.e., 0.45–0.025 μm) of the membrane, as this will affect the binding of larger or smaller proteins. Low pore size is recommended for low MW proteins (<20kDa), whereas for most of the proteins 0.45um pore size can be used. The binding capacity for nitrocellulose and PVDF is 80-100ug/cm2 and 170-200 ug/cm2 respectively. Hence, for low abundance proteins or low affinity antibodies, PVDF can be chosen to increase sensitivity. It is important to note that protein transfer onto PVDF membranes may be inhibited by high SDS concentrations, so prior to transfer gels should be thoroughly washed in distilled water (e.g., 2 min for thin gels), and then equilibrated in transfer buffer (e.g., 5 min) to remove excess SDS. PVDF are good choice for detecting hydrophobic and transmembrane proteins. Electroblotting is the most commonly used procedure to transfer proteins from a gel to a membrane. The main advantages are the speed and the completeness of transfer. Electroelution are two types (a) complete immersion of a gel membrane sandwich (wet transfer) or by (b) placing the gel-membrane sandwich between absorbent paper soaked in transfer (semi-dry transfer). The semi-dry transfer has an advantage of being faster and requires small volume of transfer buffer. Whereas wet transfer offers better transfer efficiency specially for larger proteins as well as higher and consistent quality of transfer. Utilizing the same principle as PAGE, the negatively charged proteins in the gel are transferred across onto the membrane when a lateral electric current is applied while immersed in a buffered solution. The membrane is placed directly upon the gel ensuring a mirror image transfer of proteins occur. The composition of the transfer buffer also plays an important role in the efficiency of transfer, especially for proteins of a high molecular weight. The common transfer buffer formulation is 25 mM Tris-HCl pH8.3, 192 mM glycine, 20% (v/v) methanol or ethanol. SDS is not included in transfer buffer as SDS present in proteins after gel transfer is sufficient to move them to membrane. It is important to note that the buffer pH should not be adjusted through the addition of acidic or basic solutions, as this will result in higher conductivity through greater ion content, thus increasing the temperature of the solution and potential background interference and/or inefficient transfer. These buffers may be pre‐chilled to prevent overheating, due to the high current, and the deformation of the membrane, with transfer tanks commonly containing ice blocks. Methanol or ethanol in the transfer buffer helps to increase the ability of membranes to bind proteins and prevents gel swelling. However, inclusion of methanol within these buffers may decrease the transfer of larger molecular‐weight proteins (>100 kDa) as it reduces the pore size of within gels. Hence, it is advisable to exclude methanol when probing for larger proteins. For the majority of proteins analyzed via western blotting technique, low‐ionic strength buffers and low electrical currents are optimal. If these generic rules are followed, then successful transfer is highly likely. To transfer proteins, a typical sponge-paper-gel-membrane-paper-sponge transfer sandwich is assembled. After adding membrane it should be checked that there are no air bubbles. The gel first immersed in transfer buffer and after adding membrane on top of the gel a roller should be rolled to take out any trapped air bubbles. The sandwich after closing is clamped with the transfer cassette carefully and then immersed in to the transfer buffer. The proteins are negatively charged due to SDS left from the SDS0PAGE gel and will travel from negatively charged cathode toward positively charged anode when an electric field is applied. A typical setting for transfer is 100 V for 1hr. However, for high molecular weight proteins constant 0.4 A for 2 hrs should be used, concomitant with the immersion of tank into ice-water bath. Gel staining allows the evaluation of transfer efficiency, through visualization of protein remaining within a gel (e.g., using Coomassie/silver/Ponceau stains or stain‐free gels. Most PDF membranes cannot be effectively stained by PonceauS and instead UV transilluminator can be used to visualize proteins after transfer. Nitrocellulose membrane are easily stained by Ponceau S and they should only be submerged in transfer buffer prior to use. It is good choice for binding glycoproteins and for detection using fluorophore-conjugated antibodies. Protein visualization after transfer is done by incubation membrane in Ponceau S dye 0.1% w/v in 5% acetic acid. The background is destained via several tandem rinses with distilled water. Ponceau S staining is also good visual aid for accurate membrane cutting. Unequal loading will be displayed as noticeable differences in multiple protein band(s)/lane(s) intensities. In this instance the variability may be due to unequal sample protein quantities caused by different sample concentrations, or mistakes in the sample volume loaded into individual lanes. Protein lysate concentrations should be re-analyzed to determine if it was the former. Without efficient and complete transfer of proteins, accurate quantitation and analysis will be compromised. The dye can be completely removed by briefly washing the membrane in Tris-buffered saline-Tween 20. Once blotting is done the next step is blocking. Blocking is the stopping or arresting of nonspecific antibody interaction. Many blocking agents are present in the market ex non-fat dry milk, 3-5% bovine serum albumin, etc.
Blocking – After transferring of proteins and Ponceau staining and cutting membrane, it needs to be subjected to a blocking step. The blocking step reduces nonspecific binding of primary/secondary antibodies. Blocking solution precisely binds to sticky sites on the membranes, thus preventing the antibodies from binding non-specifically to those sites. As membranes have a high affinity for binding proteins and therefore antibodies blocking reduces background in subsequent steps. Different solutions may be used for this stage of the protocol, each having their own benefits and limitations. The most popular blocking solutions are 5-10% non-fat milk or 3-5%BSA dissolved in Tris Buffer Saline Tween‐20 (TBST). Non‐fat‐dried milk diluted in TBST is often used and is cheap and widely available, and milk proteins are not, however, compatible with all antibodies. For example, milk contains phosphor-casein, which may interfere with certain phospho-specific antibodies. The interference might increase the background/noise and/or decrease the signal. While working with streptavidin-biotin detection method milk shouldn’t be used for blocking, as milk contains biotin and will interfere with the detection. In the preparation of blocking solution, the mixing must be thorough and the solution should then be filtered to prevent grains contaminating the blot during development. BSA is a safe blocking agent for most phosphor-specific antibodies but it cannot be used when lectin probes are used, as BSA contains carbohydrates. A further (albeit less common) option for blocking is using highly purified non‐animal proteins (e.g., isolated casein), although these are much less cost‐effective. Usually, commercial companies provided proper guidance on blocking conditions. However, as discussed in previous para, not all blocking conditions are suitable for all target proteins. Hence, validation and testing for each individual protein of interest are always recommended. Whichever approach is chosen an optimal blocking buffer should improve the sensitivity of the western blotting by reducing background and improving the signal‐to‐noise ratio. The signal to noise ratio is measured as the signal obtained in a sample containing the target analyte vs that obtained with a sample without the target analyte. Ideally, a blocking agent will bind to all sites of non‐specific interaction, eliminating all background without altering access/interaction to the protein(s) of interest for antibody binding. The choice of blocking agent should be based on the antigen itself and the type of secondary Ab conjugation. PBS interferes with phosphatases hence in the assays where alkaline phosphatase conjugates are used TBST should be selected. PBS interferes due to the presence of sodium phosphate (and in some solutions potassium phosphate also). If PBS is to be used for intermediate steps, the membrane should be sufficiently washed in TBST to remove excess sodium phosphate before addition of the substrate. A good western blotting outcome depends not only on the choice of blocking agent, the volume of blocking agent and indeed the incubation period for blocking (which may vary from an hour to overnight) are also important factors. Too little blocking agent (low concentration) or too short an incubation period will increase the potential for non‐specific binding of the primary Ab to the membrane‐bound proteins which could result in the excessive background and/or reduced signal‐to‐noise ratio. Conversely, an incubation period that is too long and/or uses excessive blocking agent may interrupt antigen‐antibody interactions, also causing a reduction in signal‐to‐noise ratio. A comparative study was reported where authors assessed the efficacy of commonly used blocking protocols to minimize non‐specific background and promote immunoreactivity of antibodies (monoclonal, polyclonal or biotin‐conjugated) against phospho‐amino acids. The authors recommended the use of a solution composed of 5% BSA, 5% Amicase and 5% membrane‐blocking agent in Tris‐buffered saline containing 0.05% Tween20 for 45 min at room temperature to achieve good‐quality low background. Amicase is a Sigma product which is a mixture of free amino acids with virtually no unhydrolyzed peptides and minimal inorganic components. Tween 20 aids in the removal of residual SDS from earlier steps. As with most common blocking agents, incubation for longer than 1.5 h led to reduced signal strength (signal to noise). While a 30 min or less incubation resulted in unacceptably high backgrounds. As no single blocking agent is ideal for every western blotting assay as each antigen‐antibody pairing has unique characteristics. Keeping the above considerations in mind the chosen blocking strategy must be optimized for each individual application. Most commonly, the transferred protein is then probed with a combination of antibodies: one antibody specific to the protein of interest (primary antibody) and another antibody specific to the host species of the primary antibody (secondary antibody).
Incubation – Primary antibody: It is the final step in the western blotting technique. This probing process is carried in two steps. The primary antibody has the binding capacity with the protein of our interest. The primary antibody is probed into the blotting membrane. Antibodies also known as immunoglobulins are naturally produced by the immune system in response to antigens. Antibodies are immune system-related proteins called immunoglobulins. Each antibody consists of four polypeptides– two heavy chains and two light chains joined to form a “Y” shaped molecule. The chains are connected by disulphide bonds. Reducing agents such as DTT and b-mercaptoethanol are required to completely denature them. Each tip of the “Y” of an antibody contains a paratope (analogous to a lock) that is specific for one particular epitope (similarly analogous to a key) on an antigen, allowing these two structures to bind together with precision. Primary antibodies used for western blotting are often produce in animal hosts such as rabbit, mouse, rat, goat, sheep etc. They can be developed to not only recognise a protein but also specific modification, such as phosphorylation, acetylation, glutathionylation, glycosylation or methylation. The principle of the western blotting is the detection of protein(s) through the binding and recognition of antibodies (Ab) to one or more targets. This interaction has to be highly specific between a portion of the antigen (protein) or epitope and the specific recognition sites found on the fragment antigen‐binding (Fab) region of the antibody termed a paratope. The primary Ab should be thoroughly assessed and validated to be specific and sensitive enough to detect the intended target protein. Specificity and performance of the primary Ab antibody is also dependent on whether it is monoclonal (mAb) or polyclonal (pAb) and both have disadvantage/advantage. Polyclonal antibodies are produced from differing B‐cell lineages, recognizing multiple epitope regions on an antigen. Polyclonal antibodies are cheaper, easier and quicker to generate. They also provide an extra signal amplification step, since more than one antibody molecule from a polyclonal pool will bind per protein, enhancing sensitivity of western blotting. Also, they are better reagents for immunoprecipitation. Sometimes Polyclonal antibodies have major variations, mainly due to differences in the mix of antibodies generated by the animal. However, not only each host animal will have a different quality polyclonal antibody batch, but also different serum bleeds from same host will have a varying level of quality. Hence, the disadvantage with polyclonal antibody is a limiting amount that can be obtained from each batch and a significant batch-to-batch variation. Monoclonal antibodies are advantageous compared to polyclonal due to similar specificity of each batch of monoclonal from the same hybridoma. It is important to check that the antibody is specific toward the native or denatured protein. Because antibody binding affinity may get affected due to the denaturing treatment of protein samples prior to SDS‐PAGE. This may alter the exposure and availability of the epitope. In some cases, it may be necessary to use “native‐specific” monoclonal antibodies. Monoclonal antibodies are ideal tool for western blotting since high affinity clones are usually selected during the antibody generation process. Moreover, they are relatively easy to produce at high quantities, providing a reliable source of consistent high quality reagent, with little or no variation between batches. Although monoclonal antibodies are superior to polyclonal antibodies with respect to being able to generate the same antibody in different batches, monoclonal antibodies also have problems. Sometimes hybridoma cell lines can be contaminated and not grow after storage or may die off. One possibility to overcome some of the limitations of monoclonal and polyclonal antibodies is the use of recombinant antibodies. Modern approaches utilize synthetic peptides, often producing Ab toward short denatured 8–10 amino acid sequences. Several projects are currently underway to improve the quantity of renewable validated antibodies available. In several ways recombinant antibodies are superior to monoclonal and polyclonal. They have the ability to engineer the antigen binding site, the ability to rapidly produce many variants to a specific antigen and the ability to quickly screen these variants. However, the main advantage of recombinant antibodies is that it can be defined by its sequence, and researchers anywhere would be able to create the same recombinant antibodies, significantly reducing the lot-to-lot variability. If a batch of recombinant antibody is contaminated or lost, new ones can be created if the antibody sequence is known. However, like all antibodies, validation of recombinant antibodies is needed before it should be used for western blotting. Affinity chromatography and/or anion‐exchange filtration are utilized generally for isolating antibodies and small proteins depending on the class. Predicted and confirmed species cross‐reactivity information is often only available through the vendor; however, binding will entirely depend on the antigen region. When choosing a primary Ab, there may be multiple forms available from different vendors. Ideally each would bind to a unique antigen upon the protein of interest, allowing accurate assessment of the protein’s abundance. However, this is often not the case, and assessment of the previous literature utilizing that antibody is strongly advised. Some proteins may be orthologous and contain the same or similar sequence to other species. Assessment of new antibodies for a target antigen requires even more careful testing, including the use of a variety of appropriate positive and negative controls. Validating an antibody is different from characterizing an antibody. Importantly, validating an antibody doesn’t just involve determining the antibody’s specificity for its target but also involves determining its reproducibility. Reproducibility demonstrates that the antibody’s properties are independent of lot-to-lot variations. Methods for validating antibodies for western blotting do not apply to other applications utilizing the same antibodies including immunohistochemistry and enzyme-linked immunosorbent (ELISA) applications. Primary antibodies are best diluted in 3-5% BSA in TBST. However, certain antibodies may require to be diluted in milk to reduce the non-specific background. All antibody solutions should be supplemented with 0.005% sodium azide, in order to inhibit microorganism growth and increase the shelf life. For the final working concentration for antibodies, 2-3 fold lower concentrations work. The optimal concentration depends on cell type and amount of protein loaded. A really high concentration of antibody can result in high background whereas too little will give faint bands. Incubation of antibodies can increase the specific binding to the target protein and lower the background noise. Increasing temperature of incubation temperature to 37ºC also shortens the incubation time to 30 mints.
Secondary antibody: Next the secondary antibody is attached to the part of the primary antibody. This secondary antibody has identification characteristics. Multiple secondary antibodies bind per primary antibody providing a crucial signal amplification step, contributing to the sensitivity of western blotting. They are either conjugated to an enzyme such as horseradish peroxidase (HRP), alkaline phosphatase (ALP), or to fluorescent probes. The secondary antibody dilutions range is high compared to the primary antibody. It ranges from 1:1000 to 1:20,000 depending upon the brand, target protein abundance, and primary antibody affinity. They can be diluted in TBST or blocking buffer. Different antibodies require different storage hence it becomes imperative to check for proper storage conditions. Some need -80ºC, some -20ºC whereas few need 4ºC. It is important to aliquot into single use volumes to avoid freeze thaw cycles. Antibodies conjugated to enzymes shouldn’t be frozen as enzyme activity gets effected. Also, conjugated antibodies shouldn’t be be supplemented with a solution containing azide as it will completely inhibit HRP and result in complete loss of signal. Alternatively, fluorescently-tagged antibodies can be used, which require detection using an instrument capable of capturing the fluorescent signal. Fluorescent blotting is a newer technique and is growing in popularity as it affords the potential to multiplex (detect multiple proteins on a single blot). Whatever system is used, the intensity of the signal should correlate with the abundance of the antigen on the membrane.
Blot washing – The standard TBST formulation (50mM Tris-HCL pH7.5, 150mM NaCl, 0.1% tween 20) is stringent enough for most antibodies. Before incubating with secondary antibody In order to remove unbound primary antibodies and secondary antibodies before detection step blot needs to be washed 3-4 times with TBST for 5-10 min each wash. However, NaCl and tween 20 concentrations can be increased 1.5-2 fold in order to decrease background and increase specificity.
Detection – Often the secondary antibody is complexed with a label, which when combined with an appropriate substrate, will produce a detectable signal. Labels (or conjugated molecules) may include biotin, fluorescent probes such as Invitrogen AlexaFlour or DyLight flourophores, and enzyme conjugates such as horseradish peroxidase (HRP) or alkaline phosphatase (AP). Chromogenic substrates produce a precipitate on the membrane resulting in colorimetric changes visible to the eye. The most sensitive detection methods use a chemiluminescent substrate that produces light as a by-product of the reaction with the enzyme conjugated to the antibody. The light output can be captured using film. However, digital imaging instruments based on charge-coupled device (CCD) cameras are becoming popular alternatives to film for capturing chemiluminescent signal. Detection of the protein of the interest can be done colorimetric, chemiluminescence or radiography methods. Procedures vary widely for the detection step of a western blot experiment. One common variation involves direct versus indirect detection. With the direct detection method, an enzyme- or fluorophore-conjugated primary antibody is used to detect the antigen of interest on the blot. This detection method is not widely used as most researchers prefer the indirect detection method for a variety of reasons. In the indirect detection method, an unlabelled primary antibody is first used to bind to the antigen. The indirect method offers many advantages over the direct method. The general principles of detection for western blotting are the same as for other antibody‐based assays, such as the enzyme‐linked immunosorbent assay (ELISA). The two most commonly employed enzymes are alkaline phosphatase and horseradish peroxidase. Both alkaline phosphatase and horseradish peroxidase can be used for colorimetric or chemiluminescent detection. When employing horseradish peroxidase, it is important to remember that sodium azide inhibits this enzyme so solutions like those used for blotting and detection should not contain sodium azide, or membranes should be sufficiently washed before exposure. In colorimetric a substrate (e.g., 3,3′‐diaminobenzidine) of HRP is oxidized producing a brown insoluble product. Quantification can then be achieved by scanning the blot with either a dedicated imager or traditional office scanner and analyzing band intensity using software. A key disadvantage of this form of detection is that the chemical reaction must be stopped, and therefore, optimal reaction conditions need to be determined (reaction time, temperature, etc.) prior to quantitation. With chemiluminescent detection, HRP luminol is oxidized in the presence of hydrogen peroxide producing 3‐aminophthalate, which emits light at 425 nm. A chemiluminescent blot is optimally imaged after 3–5 min of incubation with the substrate and may produce a signal for several hours. A key advantage of this form of detection is that the blot can be repeatedly rinsed and exposed to substrate and luciferase to allow multiple exposures. This is useful for optimization of detection parameters, thereby ensuring the blot is not underexposed or overexposed. However, a potential disadvantage is that the blot must either be exposed to film, which must then be developed, or be placed in a specialized detection system with the latter being the most common and accurate approach. Both options for visualization of the blot, however, require more expense and equipment than for colorimetric detection. Quantification of the blot will be dependent on the light detection method. If photosensitive film was exposed and developed, subsequent scanning and Image J are commonly employed. More commonly, a specialized detection system, typically utilizing proprietary software, employing densitometry analysis will be used. The proper storage and age of buffers, substrates, stop solutions, and luciferase solutions must similarly be considered as degradation and/or contamination of any one of these can affect each of the steps and potentially prevent detection and/or result in very high background signals. It is also important that the secondary Ab has been properly stored (−80 °C, −20 °C, 4 °C) and is reasonably fresh, as the enzymes will degrade with time. If stored correctly, antibodies may still be viable for multiple years. However, suppliers only recommend storage for approximately a year as extended storage will decrease its effectiveness. These aforementioned limitations can be overcome using fluorescent detection where the secondary Ab is conjugated to a fluorophore. For this type of detection, no further chemical reactions are required, and the blot can be visualized after exposure to secondary Ab and rinsing. However, it does require further specialized imaging systems. Fluorescent‐based imaging has a greater upper linear range of detection (250–500 pg) compared to chemiluminescent (125 pg). Fluorescent antibodies also have a similar lower range when compared to chemiluminescent detection and so may be more appropriate when investigating higher abundance proteins. A further benefit of fluorescent antibodies is the ability to utilize primary Ab from different species. Subsequently different wavelength secondary Ab for each to perform multiplexed exposures upon the same blot. This crucial difference can allow the probing of multiple targets with similar molecular weights or PTMs using different conjugated fluorophores, specific to different excitation channels. When it is desirable to improve the sensitivity for the detection of low abundance target proteins, a couple of approaches may be employed. First, switching detection systems may help as fluorometric detection is generally similar to chemiluminescent, whereas chemiluminescent detection is more sensitive and produces signal at lower concentrations than colorimetric. Second, just as using a labelled secondary Ab boosts the signal compared with using a labelled primary Ab, use of a labelled tertiary antibody can boost signal by providing additional amplification. The stripping and re‐probing of western blot membranes provides a time‐efficient method for determining multiple protein targets within a single gel run. It allows a number of different analyses on a single membrane, thus saving time, sample and consumables, and maximizing efficiency. Furthermore, there may also be times where a membrane requires probing with a primary Ab to confirm the data obtained from the initial analysis of the protein of interest (i.e., a different 1°Ab specific to another epitope on the same protein). Initially developed in the early 1980s, these methods involved incubation in either urea, β‐MCE/BSA buffers, or highly acidic glycine buffers (pH 2.2) at elevated temperatures for long periods (up to an hour in some cases), in order to remove the 1°Ab and detection reagent (e.g., enhanced chemiluminescence [ECL]). Ultimately, multiple protein targets could be detected within a single sample/run, since the 1°Ab have been disassociated from their target proteins, allowing other 1°Ab to be added. Since these early experiments, stripping buffers have progressed rapidly and many commercial preparations exist which claim to be gentler (generally consisting of a mixture of SDS, glycine, and detergents; TBST) and work rapidly (<15 min) at room temperature. However, it is always recommended to check the efficiency of these commercial buffers, by re‐exposing the membrane with ECL reagents or other detection methods post removal of the antibody.
Quantification – It is very important to be aware that the data produced with a western blot is typically considered to be semi-quantitative. This is because it provides a relative comparison of protein levels, but not an absolute measure of quantity. There are two reasons for this; first, there are variations in loading and transfer rates between the samples in separate lanes which are different on separate blots. These differences will need to be standardized before a more precise comparison can be made. Second, the signal generated by detection is not linear across the concentration range of samples. Thus, since the signal produced is not linear, it should not be used to model the concentration.
Stripping and re‐probing are commonly undertaken to investigate the levels of phosphorylated proteins before assessing the total expression of the same protein allowing the signaling capacity vs activation to be assessed (discussed below). In doing so, the generally weaker expressed phospho‐protein is first detected, allowing accurate measurement, before the total is probed. Despite the obvious benefits of stripping and reprobing of membranes, these methods are not always foolproof and can have significant limitations. First, care must be taken as to which protein the membrane is re‐probed for the following stripping, as it is difficult to eliminate detectable signal from highly abundant proteins. Additionally, if the stripping buffer has failed to completely remove all antibody and ECL from the membrane, and the two proteins of interest are of similar molecular weights [e.g., GAPDH (molecular weight: 35.8 kDa, but band commonly detected at 37 kDa) and ERK 1/2 (molecular weights: 42/44 kDa, respectively)], then the signal from the ineffectively stripped antibody may interfere with the subsequent detection and quantification of the second target protein. Further to this, stripping of the membrane cannot be performed indefinitely. As a rule of thumb, only three stripping incubations are recommended, due to loss of the antigen. This limits the number of targets that can be probed from the same membrane but is still worth doing considering the benefit of measuring two or more targets within the identical sample. There are, however, a number of recent developments which have aimed to enhance the capacity for multiple Ab detection from the same membrane. In 2009 Sennepin et al. described a technique whereby instead of removing or stripping the HRP activity linked to the Ab from the membrane, this is instead irreversibly inhibited with hydrogen peroxide, to allow up to five different sequential incubations/detections to be performed on the same membrane. There are suggestions, however, that the exposure to such strong oxidizing conditions may alter certain epitopes, potentially affecting Ab recognition. A similar procedure can also be performed using sodium azide, which has been proposed to avoid this epitope issue; however, the method requires lengthy incubations of upwards of 16 h compared to 15 min with hydrogen peroxide, and as sodium azide is known to inhibit HRP detection, it may interfere with subsequent detection. As previously discussed, both colorimetric and chemiluminescent approaches may utilize HRP‐conjugated 2°Ab, and both may be utilized separately in the same blot for different targets. Initially, the weaker target is blotted for using colorimetric 3,3′,5,5′‐Tetramethylbenzidine detection before heating in a β‐MCE containing buffer before re‐probing for the additional target with ECL detection.
Care should be taken with these methods and conditions should always be optimized; in addition, it should also be demonstrated that stripping and re‐probing do not adversely influence subsequent quantification. Thus, the undertaking of stripping and re‐probing must be carefully considered as the probable loss of an antigen may, in fact, lead to erroneous measurements. This becomes more important when changes in protein expression are small or gradual (i.e., time‐course experiments). However, the ability to measure both the phosphorylated and total expression of the same protein within the same blot is extremely desirable.
Analysis
As many imaging systems require proprietary software for image acquisition and quantification hence analysis of the bands of interest depends on the type of detection and the imaging system available. Each software package utilizes slightly different methods for quantification. Generally, peak height or area is used for quantification. During quantification, it is essential to ensure that the bands of interest are within the linear range of detection, as pixel saturation on imaging sensors may occur within highly abundant targets. This being a common error, can easily be avoided. However, automatic detection of saturation is now a standard feature within most imaging packages. If oversaturation occurs, it may be possible for the sensitivity of the camera to be reduced (i.e., reduced pixel binning) increasing the image resolution (larger image, more pixels) and therefore requiring more signal to achieve oversaturation. Despite this, oversaturated “staining” of the membrane (i.e., as signified by visible yellowish bands on the membrane prior to exposure) during chemiluminescent detection by highly abundant proteins (e.g., GAPDH) may occur requiring the reduction of either sample loading or greater dilution of the primary Ab. Another important consideration for quantification is the choice of single band or the whole lane boxed analysis. The former suitable for individual protein analysis, and the latter better for the detection of global protein changes (i.e., ubiquitinated proteins or puromycin incorporation). The Single-band analysis is undertaken simply through identifying sample lanes before determining the band of interest. Whereas whole lane analysis will measure the intensity within a designated area (i.e., single lane). Whichever method is chosen, it must be consistent throughout the blot, with an equal area of analysis used per lane; otherwise different volumes (size of the analysis box region) will be analyzed producing potentially distorted data. Although western blotting conditions will have been optimized to produce the clearest bands possible, a visible background may still occur, requiring background subtraction during analysis. Principally, this is achieved by subtracting the calculated background density from peak values; however, the different algorithms used to determine background intensity may not be truly representative. One common method is the rolling disk algorithm, which determines peak origins based on where a specified disk could “roll,” typically the smaller the value (smaller disk), the greater amount of background becomes subtracted. This approach is better suited to prominent individual bands since smaller, less intense bands may become lost. Another method is to manually designate a region of consistent background with no bands, assigning that density value for background correction. This method may be more suitable for whole blot analysis, with minimal fluctuation in background levels. Ultimately, the choice of analysis must be representative of the visualized blot and within the range of linear detection of the imaging method employed.
Normalization of target protein abundance
To account for possible errors in sample preparation and loading, normalization of samples to remove inter sample/gel variation is paramount. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) or β‐actin are “housekeeping” proteins (HKP). They are probed for acting as internal loading controls assuming their expression remains stable under the experimental conditions used. The HKP chosen should be one which is constant between control and experimental samples. It should also remain unaffected by the treatment or intervention undertaken. Errors such as loading more sample within one well will increase the target signal, likely skewing data interpretation. As such, target measurements may be normalized to HKP values, removing loading bias. However, the accuracy and effectiveness of these HKPs are dependent on multiple factors such as oversaturation of the protein, high background, and lack of linearity and can easily suffer from technical errors within the Western blot process. Expression of HKPs such as β‐actin has also been shown to be extremely variable between tissue types (i.e., muscle, heart, fat). Alarmingly, however, expression of β‐actin has been revealed to not be homogeneous within a single tissue sample, being shown to differ between proximal and distal regions of a single mouse sciatic nerve. Such differences in expression may become more problematic within skeletal muscle. As it is a large complex tissue, with different regions potentially responding differently to stimulation. In such cases western blot results only provide an average of expression changes within a single sample. The use of a second HKP may help; however, some of the same issues may exist and it will need to be demonstrated that the choice of HKP does not affect the interpretation. Due to the potential changes in HKP expression in response to the experiment and the limited linear range of some, the use of HKPs for normalization may mask or confound potentially relevant changes in protein expression. A viable alternative to blotting for HKPs is to assess the total amount of protein. Assessing total protein offers distinct advantages over HKPs as it is unbiased with respect to changes in the expression. If stained membranes are utilized it allows evaluation of the blotting process and transfer quality. Membranes may be stained to visualize total protein by several methods (i.e., Ponceau S, colloidal silver, India ink). However, Coomassie staining is a common, simple approach that has been demonstrated to be an unbiased method of total protein assessment. If one of these approaches is chosen, the quantification of a single random band that is consistent across each lane may be used for normalization. Coomassie staining is reliable quantification of total protein also it removes potential problems associated with individual HKP expression.
The final aspect of the analysis is the requirement to calculate the changes in protein expression, resulting from a treatment/intervention. Depending on the experimental design, multiple blots may be required to analyze all samples, and thus, subtle changes within the process may influence the final data as background or band density may be variable across multiple blots. In this instance, a single quality control sample (typically pooled from multiple controls) is loaded on each gel, providing a control sample across all gels, allowing gel‐to‐gel comparisons to be made. Dividing band values (i.e., band density) by the quality control sample (as a correction factor) normalizes differences in loading, separation, transfer, and detection that may have occurred. Another approach typically used for human studies is to use an initial basal sample for each individual to assess changes in subsequent samples, permitting the calculation of a fold change from the initial basal levels. Whichever approach is chosen, it must be applied consistently throughout the analysis and may depend on the total number of samples and comparisons needed (i.e., control vs treatment or control vs treatment 1 vs treatment 2).
Determining changes to a protein’s total expression in response to a treatment or intervention indicates its capacity to signal since a greater abundance or protein will allow a greater potential for signaling to occur. Measurements of a protein’s phosphorylation status (i.e., signaling activation) may change through modification of the individual proteins phosphorylation level or by alterations within the total amount of protein available. Consequently, the normalization of a phospho‐protein to its total expression allows the ratio of phosphorylated proteins to be assessed (i.e., the relative proportion of phosphorylated vs non). For example, within skeletal muscle, the total expression of various anabolic signaling intermediates (e.g., AKT, P70 S6K1) remained unchanged, while phosphorylation of these proteins increased. As such, the ratio of phosphorylated proteins to total expression increased, demonstrating the increased proportion of phospho‐proteins. Importantly, however, if the treatment undertaken increases both total and phosphorylation levels, this ratio may remain unchanged, masking any potential mechanisms, and thus it may be extremely important for both measures, i.e., total and phosphorylation status, to be made simultaneously.
Blots data presentation
One essential aspect and ethical concern in the reporting of scientific findings is the accurate and representative presentation of western blot data and images. Generally, western blot data are presented as both a graphical and representative image to demonstrate the effect of the intervention and the quality of blot. Herein lie ethical considerations in presenting images that accurately represent the quantified data without any modifications. For example, the splicing of images from multiple blots to form one continual image may obscure the magnitude of change between samples. If possible replicates from all experimental groups should be run within the same blot to provide a representative image; however, if this is not possible, a clear distinction between blots should be made. Additionally, sample replicates should be included in the images, demonstrating equivalent changes in response to the intervention. The inclusion of a representative loading control (i.e., Coomassie) is essential to demonstrate that changes in samples are due to the intervention, rather than protein loads. These are required to be from the same blots as the other representative images and should not be repeated unless the same gel has been stripped and re-probed for multiple targets or different molecular‐weight targets were measured. The addition of an indicator of the precise molecular weight must be included even if image cropping occurs to allow identification. For example, when blotting for phosphorylated 4E‐BP1, three isoforms typically form distinctive bands (i.e., γ, β, α) at differing molecular weights. In this instance, each isoform should be indicated with the appropriate molecular weight. Accordingly, indicators of molecular‐weight markers should be included to allow the independent determination of a target’s size and specificity. Another concern in the western blotting field is that the most commonly used normalization technique for getting semi-quantitative data is the use of “house-keeping” proteins. The best methods currently available for normalization using total protein staining utilize either Ponceau S or Bio-Rad’s Stains-free method.
Conclusions
Western blotting has emerged as an essential tool within physiological research; nevertheless, with poor understanding and implementation, any subsequent analysis can produce misleading and confusing interpretation (i.e., Ab specificity and validation). Before a sample is loaded into a gel, careful consideration must be given to often overlooked aspects such as the appropriate buffer for homogenization and extraction of the intended target protein for denaturation. Gel composition should effectively separate proteins by size, with changes to concentration giving resolution to the intended target by varying migration speed. Subsequent transfer onto an immobilizing membrane will allow the probing for one or more targets with 1°Ab and 2°Ab, with emphasis upon Ab specificity and the ability to assess PT modifications. Validation of Ab should always be undertaken, using both positive and negative controls to try to ensure specificity. Within each study design and group comparisons, QC samples should be used allowing the comparison of multiple gels. The method of detection will be ultimately be determined by the equipment available. However, fluorescent antibodies have a greater dynamic range and may be multiplexed for additional targets if desired. As sample quantity may be scarce, the ability to strip and re‐probe membranes for additional targets is desirable; however, potential issues with regard to quantification and potential signal reduction should be considered carefully and where possible mitigated. Finally, the quantification and analysis of band intensity should be evaluated consistently throughout with both single and multiple blots; as doing so can produce reliable and accurate data.
Although western blotting is a straightforward and simple method for specific protein detection, it is an error-prone method due to its time consuming multistep protocol. A major concern of western blotting is the lack of any consensus on what constitutes reproducibility. The requirement of relatively large sample amounts (usually >10 µg) makes it a difficult approach for low abundant proteins and post translationally modified proteins such as glutathionylation, phosphorylation, acetylation, and methylation. Other problems include analysis of multiple proteins from a single sample run is often cumbersome, as it usually requires stripping of membranes to remove antibody and then again re-probing with the second antibody for next target protein detection. A recent method called multiple antigen detections (MAD) was developed to allow non-stripping detection of multiple antigens on a single blot. To minimize nonspecific background in the MAD-immunoblotting procedure, the blot is incubated with secondary antibody and the membrane developed with chromogenic substrate before probing with the primary antibodies of interest. While this method seems promising, an important aspect of western blotting, the quantification of these blots, was not well described. Blots can also be cut into several pieces and used to detect targets within the molecular mass ranges of the cut membrane, but this limits information about what other targets the antibody may detect. Detection of some very large molecular weight proteins (>500 kDa) can be problematic due to difficulty in their complete transfer from gels to membranes. Moreover, improper sample preparation, proteolytic degradation of samples, lack of automation, problems with normalization of blots and quantitative analysis are other critical issues associated with western blotting procedure. The main problem associated with western blotting, Irrespective of how advanced or automated the methodology for western blotting gets is, the quality of the antibodies used. Validation of an antibody is as important as characterizing an antibody. Validation process involves determining the antibody’s specificity for its target and determining its reproducibility which means that antibody’s properties are independent of lot-to-lot variations. Methods for validating antibodies for western blotting do not apply to other applications utilizing the same antibodies including immunohistochemistry and enzyme-linked immunosorbent (ELISA) applications. Numerous examples of the problems associated with improperly validated antibodies exist. In depth, studies showed that only 6 of 53 published preclinical studies could be replicated. Due to the high production rate of new uncharacterized antibodies and the requirement of researchers to find antibodies for specific targets, it becomes difficult for manufacturers to validate all antibodies and include lot specific data. Generally, people rely on the reputation of the company or previous publications using these antibodies. However, most utilized antibodies being the best quality antibody for specific targets is not always correct. Unfortunately, currently, researchers need to independently validate key antibodies because changes in the western blotting equipment used, the temperature, the incubation times, the buffers used, or the concentrations of antibodies used may result in different results from what is provided by the antibody manufacturer. That’s reason for monoclonal antibodies being better than polyclonal as the specificity of each batch of monoclonal from the same hybridoma is the same. Polyclonal antibodies for the same immunogen raised in different rabbits show batch to batch variations, sometimes major variations, mainly due to differences in the mix of antibodies generated by the animal. Although monoclonal antibodies are superior to polyclonal antibodies with respect to being able to generate the same antibody in different batches, they also pose few problems. Sometimes hybridoma cell lines can be contaminated and not grow after storage or may die off. One possibility to overcome some of the limitations of monoclonal and polyclonal antibodies is the use of recombinant antibodies.
Over last two decades, substantial improvements in the basic western blot technique have been reported. Modifications in the preparative phase (lysis buffers, homogenization), blotting (blotting reagents, apparatus, and procedures) and detection methods have all been reported. These modifications improve the time required, the reliability, and the reproducibility of western blots.
Western blot methods and its clinical implications
The western blot technique has evolved from identification of a specific protein in a complex mixture to the direct detection of protein in a single cell. This technique has become an important analytical tool for clinical research. Western blotting is a valuable tool to study regulatory signaling processes and confirmatory serum diagnosis of HIV. A study on stem cell signaling and differentiation, as well as drug response in tumor cells, have been studied using advanced single cell western blotting technique. Also single cell western blotting could analyze cell-to cell variations in approximately 2000 cells simultaneously within complex populations of cells. With the integration of intact cell imaging, the technique allows the identification of protein expression. Western blotting and 2-DE gel separation together with the spotting of protein by peptide mass fingerprint helped to analyze clinically relevant Helicobacter pylori (H. pylori) in related gastric disease conditions (chronic gastritis, duodenal ulcer). Western blotting is commonly used for the clinical diagnosis of various parasitic and fungal diseases including echinococcosis, toxoplasmosis, and aspergillosis. The database of H. pylori (low expressed and membrane proteins) was created through the application of one-dimensional or 2-DE/MALDI-mass spectrometry techniques. In a similar manner, the Simple Western technique was employed for the analysis of 15 valent pneumococcal vaccine PCV15-CRM197. Due to its high sensitivity and automation, the Simple Western method may be extended to analyze serotypes of other polysaccharide protein conjugate vaccines. The assay was successfully used in a recent study, for the reliable serodiagnosis of Farmer’s lung disease (FLD), a pulmonary disorder caused by inhalation of antigenic particles. Thus, this technique can be exploited for rapid routine diagnosis of FLD in clinics. Different molecular and immunological methods were used for clinical diagnosis of Tuberculosis meningitis, a chronic disease of the central nervous system. However, each of these techniques has their own limitations. It was then that immunoreactivity to Mycobacterium tuberculosis antigens performed by western blotting helped in early and sensitive diagnosis. Western blotting helped to detect a new subgroup of human lymphotropic retroviruses (HTLV), in patients with the acquired immunodeficiency syndrome (AIDS). Western blotting has also been used as a test for variant Creutzfeldt-Jakob Disease and some forms of Lyme disease. It has been sometimes used as a confirmatory test for Hepatitis B and Herpes Type 2 infections. Western blots have also been used to confirm feline immunodeficiency status in cats. Recently, LD Bio Diagnostics (France) developed a commercial kit to carry out immunoblotting for the clinical diagnosis of chronic aspergillosis The commercial kit was found to be sensitive and can analyze hundreds of samples from patients with aspergillus disease. Thus, the clinical applications of western blotting technique will continue to progress as further advancements are made to improve sensitivity and reproducibility of the western blot.
Several more sensitive alternative methods such as single cell-resolution western blot, far-western blotting, diffusion blotting, automated microfluid western blotting, western blotting using capillary electrophoresis and microchip electrophoresis have all been developed to overcome problems associated with western blotting. The most recent technologies to improve the sensitivity and reproducibility of western blotting are discussed below.
Capillary and microchip electrophoresis
Few major limitations of western blotting are time-consuming nature, requirement of a relatively large amount of sample (usually 10-20 µg/assay) and detection of only one protein at a time. Moreover, it requires detection of housekeeping proteins or total proteins on the membrane to normalize the target bands detected. To address these issues, a hybrid between capillary electrophoresis for SDS-PAGE and conventional blotting for the western part referred to as capillary and microchip electrophoresis (MCE) based western blotting was developed. In brief, multiple injections of the same protein samples are loaded in separate tracks on a microchip and are captured on a PVDF membrane for immunoassay. Using only 400 ng of Jurkat cell lysate sample MCE was applied to measure 11 different proteins including ERK1/2, MEK1/2 AKT, STAT3, phosphor-ERK1, phospho-ERK2, and β-tubulin. Compared to conventional western blotting technique, MCE has a higher sensitivity with better resolution that allows measurement of multiple target proteins from a single cell lysate sample. This approach eliminates blocking steps and requires shorter analysis time (approx. 8 min for electrophoretic resolution). MCE allows the performance of parallel multiplexed assays of a group of proteins using a small sample amount.. This method is still being developed and further improvements to this technique would allow significant improvements in multiplexing protein detection.
Automated microfluidic protein immunoblotting and single cell-resolution western blotting
Another automated microfluidic protein immunoblotting technique were developed to save time, avoid multiple assay steps and limit equipment and reagents requirements. This automated protein immunoblotting is a programmable controlled technique (i.e.voltage control and pressure) that combines PAGE with blotting in one device. The technique allows the integrated assay steps (PAGE, transfer and in-gel blotting) to be viewed using an epifluorescence microscope equipped with a charge-coupled device camera. In this method photo patterning (photochemical etching) of polyacrylamide gels is done on microfluid glass devices that act as a platform to integrate the multiple assay steps. The method is rapid and was employed to detect free prostate specific antigen in the human seminal fluid sample in less than 5 min. This method is economical and lowers the consumption of reagents as the glass chips that are used are reusable after simple chemical treatments. The technique is still in process of development to further improve sensitivity and enable protein quantitation.
Single cell-resolution western blotting
It was developed to detect individual cell-to cell variations in protein expression between cells. The microdevice used for this assay consists of a thin layered polyacrylamide gel with micro wells. The micro wells in the gel layer are loaded with single cell protein samples and are lysed chemically in each well to obtain single cell lysate to be resolved on PAGE. Subsequently, the proteins are immobilized on the same gel using ultraviolet (UV) light and are probed with antibodies for immunoblotting. Multiplexing of a single cell is achieved by incubating immobilized proteins with different antibodies using stripping/ re-probing protocols. From a single cell, multiplexed data (more than 10 proteins) was obtained within a time of 4-6 hrs.
Simple Western
In 2010 a new instrument, Simple Western™, which utilized a microchip capillary electrophoresis (CE)-SDS combined with immunoprecipitation, but separated proteins based upon size instead of charge was reported. Simple is based on CE SDS where the separated proteins are attached to the wall of capillary by a proprietary photo (ultraviolet) activated chemical crosslink. Each capillary contains separation gel matrix and stacking matrix proteins are separated based upon size using high voltage. Subsequent blotting is done automatically by washing the capillary to remove the gel matrix, then incubating and washing the capillary with primary and secondary antibodies conjugated with horseradish peroxidase. The horseradish peroxide is detected by its signal generated upon exposure to luminol and an electropherogram trace or a virtual gel image displayed. The system combines immunoassay, detection, and data analysis in one machine.
DigiWest
DigiWest, is a modified version of the western blot method which increases the throughput of western blotting. The DigiWest method separates proteins by electrophoresis and transfers proteins to blotting membrane as usually done for western blots. The next step is to biotinylate
(using amino-reactive biotinylation reagents) proteins on the membrane and cut the membrane into 96 strips to create 96 different molecular weight fractions that are on the membrane. The proteins on these strips are eluted into wells in 96 well plates that contain neutravidin-coated Luminex beads. Each well contains a different distinct bead set with different color codes. After pooling the beads, the components of the entire lane are now reconstructed with different color codes corresponding to the molecular weight of the immobilized proteins. Using a small aliquot of the bead pool, new wells are then incubated with antibodies overnight and then phycoerythrin-labelled secondary antibodies added to detect the primary antibodies used. Detection of the phycoerythrin is done using a Luminex FLEXMAP 3D instrument. The advantages of this method are the increased throughput of the samples, lower amounts of lysate require/target detection, and lower amounts of antibody required.
Micro-loader
This method involves a funnel-like structure sample micro-loader device to load samples. The device is attached to the top of polyacrylamide gel and filled with 4% stacking gel solution through the outlet of the tips via capillary action. The protein in a sample that travels through the transfer pipette is concentrated by electrophoresis. The incorporation of micro-loader device in gels improved both protein separation and resolution. This technique was able to detect number of proteins and phosphoproteins in each sample by loading only 1.5 µg of protein per lane. This technique has the advantage of being relatively simple to do and would be very useful to measure protein expression and phosphorylation in samples that are limited.
Thin-film direct coating with suction-western blotting (TDCS)
TDCS is a capillary-tube based approach designed to reduce antibody consumption and time required for western blotting. This is a highly sensitive and rapid detection method for quantitative
analysis of multiple antigen-antibody interactions. The operational time for TCDS is much shorter (about 5 min) than conventional western blotting with increased signal-to noise ratio. In this method proteins are resolved on SDS-PAGE by electrophoresis and are transferred to PVDF membrane for 1 hour using 10 mM 3-(Cyclohexylamino)-1-propanesulfonic acid (CAPS) transfer buffer (pH 11) with 15% methanol. The PVDF membrane is placed on a stage and allowed to air dry for 10 min. Thereafter, the primary antibody solution (0.1 -0.2 µl) is added to the coater (super light-and-slim slot die coater or capillary tube) mounted on a translational stage for coating the membrane via capillary force/liquid pump. The coating process is less than one minute and is programmable. The gap between the coater and the PVDF membrane is controlled. The PVDF membrane is then incubated for 2-10 min followed by subsequent washing steps for less than one minute with suction (~30 cm Hg) and again coated with secondary antibody using a similar coating process. Thus, this technique is fast and enables quantitative analysis of multiple protein interactions
Troubleshooting
Even though the procedure for western blot is simple, multiple steps involved make it an error prone procedure that can lead to unexpected results. The problem can be grouped into five categories:
(1) unusual or unexpected bands,
(2) no bands,
(3) faint bands or weak signal,
(4) high background on the blot, and
(5) patchy or uneven spots on the blot.
Unusual or unexpected bands can be due to protease degradation, which produces bands at unexpected positions. In this case, it is advisable to use a fresh sample kept on ice or alter the antibody. If the protein seems to be in too high of a position, then reheating the sample can help to break the quaternary protein structure.
Blurry bands are often caused by high voltage or air bubbles present during transfer. In this case, it should be ensured that the gel is run at a lower voltage and that the transfer sandwich is prepared properly.
Non flat bands can be the result of too fast of a travel through the gel, due to low resistance. To fix this the gel should be optimized to fit the sample. Finally, white (negative) bands on the film are due to too much protein or antibody.
Another problem: no bands can also arise due to many reasons related to antibody, antigen, or buffer used. If an improper antibody is used, either primary or secondary or if the concentration of the antibody is not appropriate. if the concentration is too low, the signal may not be visible. Another reason for no visible bands is the lowest concentration or absence of the antigen. In this case, an antigen from another source can be used to confirm whether the problem lies with the sample or with other elements, such as the antibody. Moreover, prolonged washing can also decrease the signal. It should be ensured that all buffers are fresh and noncontaminated.
Weak signals can be caused by the low concentration of antibody or antigen. Increasing exposure time can also help to make the band clearer. Another reason could be non-fat dry milk masking the antigen. In this case, use BSA or decrease the amount of milk used.
The high background is often caused by the too high concentration of the antibody. Increasing the washing time can also help to decrease the background. Additionally, too high of an exposure can also lead to this problem. Therefore, it is advisable to check different exposure times to achieve an optimum time.
Patchy and uneven spots on the blot are usually caused by improper transfer due to air bubbles trapped between the gel and the membrane. It is also important to use a shaker for all incubation to avoid uneven agitation during the incubation. Filtering the blocking agent can also help to remove some contaminants. Finally, this problem can also be caused by aggregation of the secondary antibody; in this case, the secondary antibody should be centrifuged and filtered to remove the aggregated.