Antibodies are members of immunoglobulin family. They constitute 20% of the plasma protein. Different population of antibodies are present in different parts of the body. They are host proteins produced in response to foreign molecules. This is the body’s self-defence mechanism against foreign bodies. B- Lymphocytes in the body have cell surface receptors for foreign bodies. On attachment of these bodies, they differentiate to form lymphoid or plasma cells, which in turn produce antibodies.
Science has paved way for the understanding of antigen-antibody interaction and their characteristics. Now scientist can manipulate these characteristics of antibody fragments for different usage. There are many application of antibodies in diagnosis and therapy. The use of recombinant technology opens up the potential to create an infinite number of combinations between immunoglobulins. We can now manipulate these fragments to our advantage. As the antibody usage has increased, our understanding about these molecules has increased. Various methods of purification technique have been identified and used for commercial applications. All these applications require antibody in a purified state. The state of purity depends on the scale of application. The parameters for purification entirely depends on its intended application. The parameters can be physical such as size, charge, pI, stability. Purity can be a scale from microgram to a gram or any other weight measurement.
Structure of Immunoglobulins
All immunoglobulins have a common structure with four polypeptide chains; two identical heavy (H) chains, each carrying covalently attached oligosaccharide groups; two identical, nonglycosylated light (L) chains. A disulfide bond joins the heavy chain and the light chain together. The heavy chains are also joined by disulfide bonds. The disulfide bonds are present in the flexible region of the heavy chain known as hinge region. All four polypeptide chain s contains constant (C) and variable (V) regions found at the carboxyl and amino terminal portions, respectively. All chains have a single V region. Heavy region contains 3 C region and light regions have a single C region. The V regions combine to form two identical antigen binding sites.
Immunoglobulins are divided into five major classes according to their H chain components: IgG, IgA, IgM, IgD, and IgE. The light chain molecules have k chains and l chains. Individual molecules have either one of the molecules.
Antibody fragments
Partial enzymatic digestion of immunoglobulins generates biologically active antibody fragments. These fragments can also be produced using recombinant technology.
The most common types of antibody fragments are:
Fab and Fc fragments: Digestion by papain digestion creates three molecules. Two antigen binding fragments (Fab) and one crystallizable fragment (Fc).
F(ab’)2 fragment: Digestion by pepsin creates 4 molecules. One hinge, two Fab units and a fragment containing two antigen bonding sites.
Fv fragment: an unstable fragment able to bind to an antigen. An Fv fragment has two V regions, VL and VH. Single chain Fv fragment (scFv): scFv is a stable variant of Fv, commonly produced by recombinant technology, in which a peptide linker connects the two V regions.
Dabs: domain antibodies, the smallest functional entity of antibodies.
Fd fragment: the N-terminal half of the H chain.
Polyclonal antibodies
A host will produce a large number of antibodies that recognize independent epitopes on the antigen. The serum is the source of polyclonal antibodies.
Monoclonal antibodies
Monoclonal antibodies (MAbs) are highly specific antibodies. They are produced from hybridoma cells. These cells are produced by isolating plasma cell precursors and fused with immortal cells.
Antibody sources and their associated contaminants
Antibodies and antibody fragments are produced from a variety of native and recombinant sources. The choice of source material can affect the selection of techniques for sample preparations and purification protocol. Each source contains specific contaminants associated with them. Human serum may contain albumin, transferrin, macroglobulin and other serum proteins as contaminants. Hybridoma, cell culture supernatant, may contain phenol red, albumin, transferrin, bovine IgG, other serum protein, viruses. Recombinant antibody sources may contain proteins from the host microbes, hamster ovary, etc,.
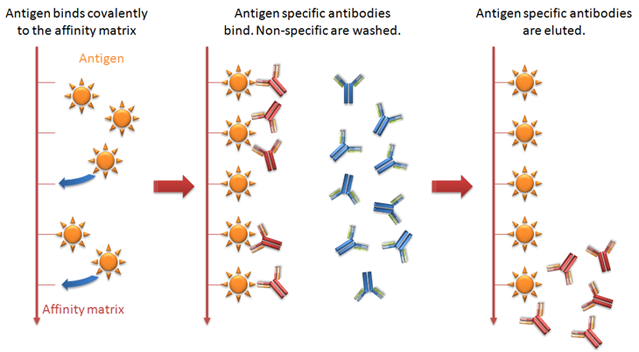
Antibody purification by affinity chromatography: Small scale
Specifity of the antibodies finds its application in this purification process. Affinity chromatography is often the first step in purification for many antibodies. Sometimes a polishing process is required after affinity process to get the required homogenicity. Affinity purification offers high selectivity. More than 95% purity can be achieved in this method. In most applications this level of purity is desired.
Affinity ligands for antibody purification
Protein A and Protein G
High affinity of these two proteins for the fc region of polyclonal and monoclonal IgG type antibodies forms the basis of purification of IgG, IgG fragments containing the Fc region and IgG subclasses. Protein G and protein A are bacterial proteins from group G streptococci and staphylococcus aureus, respectively. These proteins when coupled with sepharose forms a very good media for purification of antibodies. IgG class antibodies and fragments are purified by this media. Binding strengths obtained from individual proteins can be used as guidelines in purification process. Single-step purification of sample from native sources will purify host IgG and trace amount of proteins. In cases like this immunospecific affinity using anti-host IgG native bodies is coupled to media for effective seperation. Other techniques like Ion Exchange chromatography and hydrophobic interaction chromatography can also be used.
Protein L binds to Fv of kappaight chain
Protein L is first isolated from the surface of bacterial species Peptostreptococcus magnus. It binds with immunologlobulins through the light chains interaction. Protein L binds with a wide range of antibody classes. It binds with all antibody classes.
Ligands that bind with Fc of Fab kappa or lambda light chain
Recombinant protein of MW 13000 is commercially available in market. They bind with both light chain regions. It is produced in S. cerevisiae.
Optimization of parameters
Some parameters for antibody purification can require optimization to obtain the optimal result. Pretreatment, buffer solution, quantity of antibody to be purified, number of washes are some optimizable parameters.
Purification using Protein G Sepharose media
Protein G is generally used for the media as it shows a greater affinity for most IgG from a lot of eukaryotic species. Protein A produces highly purified antibodies. The binding strength of protein G for IgG depends on the source species and subclass of the immunoglobulin. The dynamic binding capacity depends on the binding strength and also on several other factors, such as flow rate during sample application. If harsh elution system are used leakage of ligands may also happen. Thus protein G attached at multiple points shows a very less leakage over a wide range of elution condition. Polishing is done by SEC (Size Exclusion Chromatography) and IEX (Ion exchange chromatography). Protein G can be operated in a wide range of pH. Affinity strength is maximum at physiological pH and ionic strength. Avoid excess washing, if ligand is weak.
- Equilibrate all the material used in the reaction in reaction temperature and de-gas the media slurry.
- Eliminate any air from the columns by flushing with the operating buffer.
- Re-suspend the slurry and pour the slurry in one single motion. Air bubbles should not enter the column.
- Open the bottom outlet of the column and set the pump to run at the desired flow rate.
- Maintain packing flow rate for at least 3 bed volumes after a constant bed height is reached. Mark the bed height on the column.
- Stop the pump and close the column outlet.
- Connect the column to a pump or a chromatography system and start equilibration.
Sample preparation
Centrifuge samples at 10000 g for 10 minutes to remove cells and debris. Filter through a 0.45
Buffer preparation
Binding buffer:0.02 M sodium phosphate, pH 7.0
Elution buffer: 0.1 M glycine-HCl, pH 2.7
Neutralizing buffer: 1 M Tris-HCl, pH 9.0
Water and chemicals used for buffer preparation should be of high purity. Filter buffers through a 0.45 μm filter before use.
Purification
- Prepare collection tubes by adding 60 to 200 μl of 1 M Tris-HCl, pH 9.0 per milliliter of fraction to be collected. pH of the solution should be 7.0.
- If the column contains 20% ethanol, wash it with 5 column volumes of distilled water. Use a linear flow rate of 50 to 100 cm/h.
- Equilibrate the column with 5 to 10 column volumes of binding buffer at a linear flow rate of 150 cm/h.
- Apply the pretreated sample.
- Wash with binding buffer until the absorbance reaches the baseline.
- Elute with elution buffer using a step or linear gradient. For step elution, 5 column volumes of elution buffer are usually sufficient. For linear gradient elution, a shallow gradient over 20 column volumes allows separation of proteins with similar binding strengths.
- After elution, regenerate the column by washing it with 5 to 10 column volumes of binding buffer. The column is now ready for a new purification. Desalt and/or transfer purified IgG fractions to a suitable buffer using a desalting column
Storage
Store in 20% ethanol at 2°C to 8°C.
Affinity of other antibodies
Protein A can interact with human colostral IgA as well as human myeloma IgA2 but not IgA1. Polyclonal IgA from pig, dog, and cat and monoclonal canine IgA have also exhibited binding affinity for protein A. Protein L has strong affinity for human IgA. Protein L has strong affinity for human IgD. Protein L has strong affinity for human IgE. IgM present in human and mouse serum binds weakly to protein A. However protein L has strong affinity for human and mouse IgM.
Making Immunospecific purification media with custom ligands
A ligand, a pure antigen, can be covalently coupled to a suitable matrix to create an immunospecific medium for purification. This methodology is particularly useful when the target molecules does not bonds or bonds weakly with protein A and G. It can also be used for removing contaminants. The ligand is coupled through its active amine group to a pre-active media. The pre-active media is hydrophilic in nature which contributes to the reduction of non-specific bonding of proteins.
Coupling ligands to HiTrap NHS-activated HP columns
The protocol below describes the preparation of a prepacked HiTrap NHS-activated HP column and a recommendation for a preliminary purification protocol. Many of these details are generally applicable to NHS-activated sepharose media. Coupling can take place within the pH range of 6.5 to 9.0 with a maximum yield achieved at around pH 8.0.
Buffer preparation
Acidification solution: 1 mM HCl (kept on ice)
Coupling buffer: 200 mM sodium hydrogen carbonate, 500 mM sodium chloride, pH 8.3 Water and chemicals used for buffer preparation should be of high purity. Filter buffers through a 0.45 μm filter before use.
Ligand and HiTrap column preparation
- Dissolve the desired ligand in the coupling buffer to a final concentration of 0.5 to 10 mg/ml (for protein ligands) or perform a buffer exchange using a desalting column. The optimal concentration depends on the ligand. Dissolve the ligand in one column volume of coupling buffer.
- Remove the top cap and apply a drop of acidification solution to the top of the column to avoid air bubbles.
- Connect the Luer adapter (or tubing if using a pump or system) to the top of the column.
- Remove the snap-off end at the column outlet.
Ligand coupling
- Wash out the isopropanol with acidification solution. Use 3 × 2 ml for HiTrap 1 ml and 3 × 10 ml for HiTrap 5 ml. Do not exceed flow rates of 1 ml/min for HiTrap 1 ml columns and 5 ml/min for HiTrap 5 ml columns at this stage to avoid irreversible compression of the prepacked medium.
- Immediately inject 1 ml (HiTrap 1 ml) or 5 ml (HiTrap 5 ml) of the ligand solution onto the column.
- Seal the column and leave for 15 to 30 min at 25°C or 4 h at 4°C*.
Washing and deactivation
Deactivate any excess active groups that have not coupled to the ligand, and wash out the non-specifically bound ligands, by following the procedure below:
Buffer A: 500 mM ethanolamine, 500 mM sodium chloride, pH 8.3
Buffer B: 100 mM acetate, 500 mM sodium chloride, pH 4.0
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer A.
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer B.
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer A.
- Leave the column for 15 to 30 min at room temperature or approximately 4 h at 4 °C.
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer B.
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer A.
- Inject 3 × 2 ml (HiTrap 1 ml) or 3 × 10 ml (HiTrap 5 ml) of Buffer B.
- Finally, inject 2 ml (HiTrap 1 ml) or 10 ml (HiTrap 5 ml) of a buffer with neutral pH to adjust the pH.
Storage
Store the column in a solution that maintains the stability of the ligand and contains a bacteriostatic agent, for example phosphate-buffered saline (PBS), 0.05% sodium azide, pH 7.2. pH stability of the media when coupled to the selected ligand depends on the stability of the ligand. Sodium azide can interfere with many coupling methods and some biological assays. It can be removed using a desalting column.
Performing a purification on a coupled HiTrap NHS-activated column
Use high quality water and chemicals. Filtration through 0.45 μm filters is recommended. Optimal binding and elution conditions for purification of the target protein must be determined separately for each ligand. The general protocol given here can be used for preliminary purification. For the first run, perform a blank run to ensure that any loosely bound ligand is removed. Samples should be centrifuged immediately before use and/or filtered through a 0.45 μm filter. If the sample is too viscous, dilute with binding buffer. Sample binding properties can be improved by adjusting the sample to the composition of the binding buffer. Perform a buffer exchange using a desalting column or dilute the sample in binding buffer.
Prepare the column by washing with:
(i) 3 ml (HiTrap 1 ml) or 15 ml (HiTrap 5 ml) binding buffer.
(ii) 3 ml (HiTrap 1 ml) or 15 ml (HiTrap 5 ml) elution buffer.
- Equilibrate the column with 10 column volumes of binding buffer.
- Sample preparation. The sample should be adjusted to the composition of the binding buffer. This can be done by either diluting the sample with binding buffer or by buffer exchange or desalting. The sample should be filtered through a 0.45 μm filter or centrifuged immediately before it is applied to the column.
- Apply the sample, using a syringe fitted to the Luer adapter or by pumping it onto the column. Recommended flow rates: 0.2 to 1 ml/min (HiTrap 1 ml) or 1 to 5 ml/min (HiTrap 5 ml)*. The optimal flow rate is dependent on the binding constant of the ligand.
- Wash with binding buffer, 5 to 10 column volumes or until no material appears in the effluent. Excessive washing should be avoided if the interaction between the protein of interest and the ligand is weak, since this can decrease the yield.
- Elute with elution buffer; 1 to 3 column volumes is usually sufficient but larger volumes might be necessary.
- The purified fractions can be desalted.
- Re-equilibrate the column by washing with 5 to 10 column volumes of binding buffer. The columns is now ready for a new purification of the same kind of sample. 1 ml/min corresponds to approximately 30 drops/min when using a syringe with a 1 ml HiTrap column; 5 ml/min corresponds to approximately 120 drops/min when using a syringe with a 5 ml HiTrap column. To preserve the activity of acid-labile IgG, add 60 to 200 μl of 1 M Tris-HCl pH 9.0 to collection tubes, which ensures that the final pH of the sample will be approximately neutral.
Elution buffers
Immunospecific interactions can be very strong and sometimes difficult to reverse. The specific nature of the interaction determines the elution conditions. Always check the reversibility of the interaction before coupling a ligand to an affinity matrix. If standard elution buffers do not reverse the interaction, alternative elution buffers that can be considered are listed below:
- Low pH (below pH 2.5)
- High pH (up to pH 11.0)
- Substances that reduce the polarity of a buffer can facilitate elution without affecting protein activity: dioxane (up to 10%), ethylene glycol (up to 50%).
Adding a polishing step after initial purification
One-step affinity purification generally achieves satisfactory purity of the target antibody. To achieve adequate homogeneity of the purified antibody, however, an additional polishing step using size exclusion chromatography (SEC) is recommended. This chapter also describes methods for removal of specific contaminants remaining from purification of IgG from native source or serum as well as cell culture.
Ion Exchange Chromatography
MAb has higher pI than most host cell proteins. That forms the basics of Ion exchange chromatography for purification. Most impurities like DNA, endotoxins, HCP flow through the column or washed away by buffer. Other approach is MAb is allowed to pass through while impurities gets attached to the protein A in column media.
Hydrophobic interaction chromatography
Many antibodies form dimers or aggregates, in particular at high expression levels. The aggregates are more hydrophobic in nature. It makes the aggregates bind more strongly to HIC media compared with the corresponding monomer. Therefore, HIC is an efficient tool for aggregate and dimer removal in flowthrough mode. Aggregates bind to the medium while the antibody passes straight through. HIC is also useful for removing HCP and endotoxins.
Multimodal chromatography media
New types of chromatography media that have a ligand with additional interaction mechanisms in combination with ion charges, commonly called multimodal media, are now also available. Capto adhere is a multimodal strong anion exchanger, and is designed for operation in flowthrough mode for the MAb. Multimodal media can also be operated in bind-elute mode for cases where a flowthrough step does not remove impurities such as antibody fragments.




