Introduction
During meiosis, genetic diversity in the offspring is achieved by homologous recombination. This process creates novel combinations of parental alleles and it drives evolution. The so formed zygote has a unique genetic constitution. Identification and studying the homologous recombination in a genome is important as its gives a sense of evolution. These tracking, also helps in identification of disease alleles in zygote. The variants in genetic codes is haplotyped. Also there is possibility of numerical and structural anomalies occurring during gametogenesis and are common in embryogenesis. The consequence of these anomalies and understanding of the mechanism is still difficult to achieve. This leads to an interest in analysis of both haplotyping and DNA copy number of human single cells, particularly human gametes, zygotes, and blastomeres of embryos.
The application of this understanding can be applied to avoid the transmission of genetic disorders and to improve the success of in vitro fertilization (IVF). Though genotyping in haploid cells is easy to readout reconstituting, the haplotype of a diploid cells is more challenging. The limitation of the diploid cells requirement of metaphase cells for Microfluidic separation of intact homologous chromosomes from a single cell and subsequent genotyping of chromosome-specific amplification products makes the application of technology in diploid cells difficult. This limitation is countered by family based haplotyping of diploid cells. But these haplotyping requires single nucleotide polymorphism (SNP) genotyping cells. These SNP cells are prone to errors. It is contributed by the fact copy-number state of the SNP is ignored and because the abundant whole genome amplification (WGA) artifacts in single-cell assays produce false homozygous and heterozygous SNP calls. Various methods for DNA copy-number profiling of single cells have been developed. The profiling methods rely on transforming probe intensities of microarrays or next-generation sequence read counts into DNA copy numbers.
Material and Methods
siCHILD
A computational method for single-cell genome-wide haplotyping and copy-number typing of the haplotypes in a cell, is siCHILD. This method allows determining the inheritance of linked disease variants. It can track down abnormities if either parental or mitotic/meiotic origin of haplotype anomalies in the cell. Five modules are developed for this method. It uses as input discrete genotype calls (AA, AB, BB), B-allele frequencies, and logR values of SNPs along with phased parental SNP genotypes. siCHILD is developed in R.
Module 1: Quality Control of Single-Cell SNP Data
In this module we first identify the substandard WGA cells. Perform a quality control on the single-cell discrete SNP genotypes and logR values. The cells are hybridized and the WGA products are analyzed. Discrete SNP genotypes are determined with data analysis software. The logR is the base 2 logarithm of the summed normalized SNP probe intensity values observed for each allele in the sample versus the expected summed intensity values derived from a set of normal samples(e.g., for a single cell the logR of a SNP is logR=log2(Rsingle cell/Rexpected).
For each SNP genotypes of single cells the rates of NoCall, allele-drop-out (ADO), and allele-drop-in (ADI) by using parental genotypes as control. The parental genotypes are derived from DNA samples extracted from white blood cells.
For instance, for a SNP with paternal AA and maternal BB genotypes, a cell of a conceptus is an obligate heterozygote AB; thus, detecting an AA or BB genotype for this SNP in the cell represents an ADO event. Similarly, the detection of an AB genotype in a cell of a conceptus for a SNP with paternal AA and maternal AA genotypes denotes an ADI event.
The quality of the WGA is not only expressed as mendelian errors in SNP. It might also reveal chromosomal DNA copy-number and copy-neutral anomalies present in the cell. Monosomy or a uniparental isodisomy across the whole chromosome might be understood as ADO, and NoCall across a full chromosome might indicate a nullisomy. To evaluate these scenarios, the pattern of occurrence across the whole genome ADO, ADI, and no call events are visualized genome wide inspection. It is also aided by supervised and unsupervised hierarchical clustering of SNPs. This clustering evaluates relatedness of cells as well as large-scale DNA copy-number aberrations within cells on the basis of SNP genotypes. Substandard SNPs clustered on unexpected branches are excluded from further analysis.
When high standard deviation (SD) in the logR values is found, the SNP is labeled as substandard WGA. However this high value may be contributed by from acquired numerical aberrations of chromosomes in the cell, due to chromosome instability of the cell type. To distinguish among both possibilities, we first determined the SD in logR per chromosome and subsequently summed these chromosome-specific SDs per cell to a single cumulative standard deviation (CSD) value per cell. For QC filtering, a mixture model of two normal distributions was fitted to the bimodal density function of the single-cell CSD values across all cells. Cells within 90% of the main low CSD distribution were retained for further analysis
Module 2: Single-Cell Haplarithmisis
Haplarithmisis uses single-cell SNP BAFs and phased parental genotypes to determine genome-wide haplotypes, the copy-number state of the haplotypes, as well as the parental and segregational origin of putative haplotype anomalies in the cell.
Haplarithmisis applies the following eight steps.
- The parental genotypes are phased via an available SNP genotype derived from a close relative.
- The informative SNP loci are identified. A SNP locus is defined informative when one parent is heterozygous and the other parent is homozygous for this SNP.
- The informative SNPs are categorized as paternal or maternal. An informative SNP is defined ‘‘paternal’’ when the father’s genotype is heterozygous and the mother’s genotype is homozygous. Similarly, an informative SNP is defined ‘‘maternal’’ when the mother’s SNP genotype is heterozygous and the father’s SNP genotype is homozygous.
- These maternal and paternal informative SNP loci are subcategorized on the basis of phased parental SNP genotype combinations. These SNP loci are labeled ‘‘P1’’ and ‘‘P2’’ for the paternal informative SNP category. In the maternal informative
- SNP category, SNP loci are labeled ‘‘M1’’ and ‘‘M2’’ accordingly. The SNP BAF values of the single cell are distributed into a paternal or maternal category according to the informative parental SNP genotypes defined in step 3 and step 4. Paternally informative single-cell BAF values are derived from those SNPs belonging to subcategories P1 and P2, and maternally informative single-cell BAF values are derived from those SNPs belonging to subcategories M1 and M2.
- The single-cell BAF values are subsequently mirrored around the 0.5 axis for those SNPs where either parent has a heterozygous SNP call BA after phasing. Therefore, if the cell inherited H1 of the father (and either H1 or H2 of the mother), P1 SNP BAFs will now have a value of 0 and P2 SNP BAFs will continue to have a value of 0.5. In contrast, when the cell inherited H2 of the father (and either H1 or H2 of the mother), P1 SNP BAFs will have a value of 0.5, but P2 SNP BAFs will now have a value of 1. A similar rationale applies to single-cell SNP BAFs in the M1 and M2 subcategories.
- Subsequently, per subcategory (P1, P2, M1, M2), these single-cell BAF values for consecutive SNPs in the genome are segmented by piecewise constant fitting . The resulting segments define the blocks of SNP alleles, derived from paternal H1 and H2 or from maternal H1 and H2, that co-occur on the same inherited chromosome, or in other words the haplotype blocks. Indeed, the loci where P1 and P2 SNP BAF segments jump from values 0 and 0.5 to values of 0.5 and 1, respectively, represent the sites of homologous recombination between the paternal H1 and H2. A similar rationale applies to M1 and M2 SNP BAF segments.
- These segments and the underlying processed SNP BAF values are visualized into two separate ‘‘haplarithm’’ plots, one for each parental chromosome. In the paternal haplarithm plot, segmented P1 and P2 profiles are depicted in blue and red, Similarly, segmented M1 and M2 are shown in blue and red, respectively, in the maternal haplarithm plot. Indeed, when the cell has acquired, for example, a duplication of a paternal H1 segment, P1 SNP BAFs have an expected value of 0 and P2 SNP BAF values have an expected value of ~0.33 across the duplication in the cell. In contrast, for the same duplication in the cell, M1 and M2 SNP BAFs have expected values of 0 and ~0.67 when maternal H1 was inherited by the cell, or values of ~0.33 and 1 when maternal H2 was inherited by the cell, respectively.
Module 3: Single-Cell Haplotyping via Discrete SNP Genotype Calls
For genome-wide haplotype reconstruction of a single cell via discrete SNP genotypes, the genotypes of both parents as well as that of a close relative (e.g., a sibling or the grandparents) are required.
In the current workflow two options are considered:
(1) if grandparental DNA samples are available, their SNP genotypes will be used to phase the parental genotypes and subsequently the cell’s genotype is haplotyped by applying phasing rules on informative SNPs;
(2) if DNA of a sibling is available, his or her SNP genotype will be applied to phase the parental SNP genotypes and subsequently the haplotypes of the single-cell SNP genotypes are determined by applying phasing rules on informative SNPs.
Because of allelic amplification bias and errors (e.g., ADO and ADI) after WGA, as well as the error-prone interpretation of SNP probe intensities by genotyping algorithms, individual SNP genotypes and thus SNP haplotype calls within a cell contain errors. To remove these random artifacts and to determine the SNP haplotype blocks within a cell, we designed a 1D median filter (1D-MF) that walks across the raw single-cell haplotypes for the informative SNPs genome wide and considers the raw haplotype state from multiple informative SNPs in a variable window.
Because 1D median filters preserve edges while removing noise, the locations of the homologous recombination sites in the reconstructed haplotypes of the cell are preserved. The 1D median filter window (Wk) for each chromosome ‘‘k’’ is defined as:
![]()
where Wk represents a chromosome k-specific window. W1 is the window specific for chromosome 1, containing 22 informative single-nucleotide polymorphic markers. nPMk is the total amount of informative single-nucleotide polymorphic markers for chromosome k (nPM1 is the total amount of informative SNPs for chromosome 1), and the division (nPMk/nPM1) is rounded to the nearest integer value.
Subsequently, the algorithm compares the single-cell haplotype blocks resulting from the 1D median filter with the raw SNP haplotypes of the cell and determines whether the majority of the SNPs (>60%) in the raw SNP haplotypes are assigned to the same allele as in the 1D-MF SNP-haplotype block. Otherwise the haplotype block from the 1D median filter is penalized and will not be deduced.
Module 4: Supervised Copy-Number Typing of Single-Cell Haplotypes by Integrating SNP logR Values with Haplarithmisis
In this module, the SNP logR values are normalized for %GC-bias and further to the disomic chromosomes identified via discrete SNP calls as well as SNP haplarithm patterns. Finally, normalized and segmented SNP logR values are interpreted via haplarithmisis for the detection of copy-number aberrations.
Raw logR values from SNP arrays are exported from GenomeStudio (Illumina) and are smoothed using a moving average (window of ten SNPs). These averaged logR values are corrected for %GC bias by a loess-fit and the corrected logR values are preliminarily normalized toward a trimmed mean of the likely normal disomic chromosomes determined by parental scores. The latter are determined on the basis of parent-of-origin values for SNPs. Whereas WGA produces artifacts randomly, leading to occasional aberrant parental scores for SNPs, true copy-number aberrations are expected to produce aberrant paternal or maternal scores consistently over many consecutive SNPs located within the anomaly. This principle is applied to identify chromosomes that are probably disomic. Paternal and maternal scores, PSk and MSk, respectively, are computed for each chromosome k:

where Pk,j and Mk,j represent the paternal and maternal parent-of-origin value of a SNP j informative for parent-of-origin analysis on chromosome k, respectively, and Sk,j has a value of 1 for each SNP j on chromosome k that is informative for parent-of-origin analysis. Subsequently, a parental relative ratio for each chromosome k was computed:
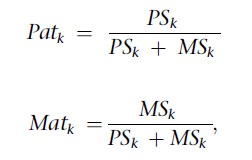
where Patk and Matk represent the paternal and maternal relative ratios, respectively. These values were used for a preliminary normalization of the logR. To fine tune the normalization, these preliminary logR profiles were integrated with haplarithm patterns, allowing a final selection of the disomic chromosomes to correct all %GC-corrected logR values of a cell according to a trimmed mean of the logR values of the selected disomic chromosomes for that cell.
To call DNA-copy-number aberrations, the segmented logR values are integrated with haplarithmisis.DNA gains and losses were scored when the segmented logR values and the haplarithm patterns across the logR anomaly were concordant. Aberrant logR values (logR < – 0.3 or logR > 0.15) not corroborated by a typical haplarithm pattern following visualization were not scored as DNA gain or loss. Aberrations smaller than 3Mb were not considered with one exception.
Genotype Inference Derived from Haplarithm Patterns
To infer discrete SNP genotypes from SNP haplarithm profiles, first transform SNP haplarithm BAF segments to discrete SNP haplotypes. Determine thresholds on segmented P1 and M1 values as well as on segmented P2 and M2 values for diploid chromosomes. These thresholds were determined by fitting a mixture model of two normal distributions to the density of the segmented P1 and M1 values, and similarly for the segmented P2 and M2 values. The distributions near to 0 and 1 were further applied (named ‘‘zone0’’ and ‘‘zone1,’’ respectively) to calculate the two thresholds—an upper threshold on zone0 and a lower threshold on zone1—which include 99% of the data in the ‘‘P1 and M1’’ and ‘‘P2 and M2’’ distributions, or zone0 and zone1, respectively. Subsequently, these thresholds were applied on the P1 and P2 segments in the paternal haplarithms as well as on the M1 and M2 segments in the maternal haplarithms. If the segmented P1 is within zone0 and the segmented P2 is not in zone1, that genomic interval is assigned the paternal H1 haplotype; however, if P2 is within zone1 and P1 is not in zone0, that genomic interval is assigned the paternal H2 haplotype. A similar rationale holds for M1 and M2 to deduce maternal discrete haplotypes. For subsequently inferring discrete SNP genotypes of the cell, the parental H1 and H2 loci determined for the cell were replaced with the respective phased parental SNP genotypes.
Optimization of Single-Cell Genotype Calling
Optimal parameters for in silico single-cell SNP typing were identified by computing the call rates and concordances of single cell heterozygous and homozygous SNPs with the expected profile determined from a matching multi-cell DNA-sample hybridized to the same platform.




