Hemoglobin –the oxygen carrier – comes in two forms: Hemoglobin is the iron-containing pigment of RBCs (red blood corpuscles) is responsible for oxygen transport. Each molecule of hemoglobin (in adults) is made of two α-globin chains and two β-globin chains. Foetuses and newborns have slightly different hemoglobin, with two α-globin chains and two γ-globin chains. At birth, the majority of hemoglobin is the fetal form. Months after birth, γ-globin production ceases and oxygen transport is performed almost exclusively by adult hemoglobin. Fetal hemoglobin is expressed in negligible quantities after the first few months of birth.
Thalassemias
Thalassemias are a type of hereditary genetic disease in which a single gene defect in either α or β globin genes results in production of abnormal RBCs and onset of anemia from a very young age. Thalassemias are further classified as Alpha-thalassemia (in which α-chains are defective) and Beta-thalassemia (in which β chains are defective). Beta-thalassemia occurs in two forms – minor and major.
Every individual inherits two copies of β-globin gene. An individual is said to have beta thalassemia minor when only one β-globin gene is defective, while the other copy is still functional. These patients are not in need of significant interventions. However, individuals with genetic defects in both copies of β-globin gene are unable to make functional copies of β-globin and suffer severe anemia within months. Beta-thalassemia major is a serious disease that varies in severity and requires treatment lifelong.
Worldwide Prevalence of Thalassemias
Thalassemias and other hemoglobinopathies tend to be most prevalent in the tropical and subtropical regions of the world. However, with increased migration, the disease occurs outside of these zones as well. Thalassemia affects around 5 in every 10,000 live births worldwide, with beta thalassemia major being the most frequent.
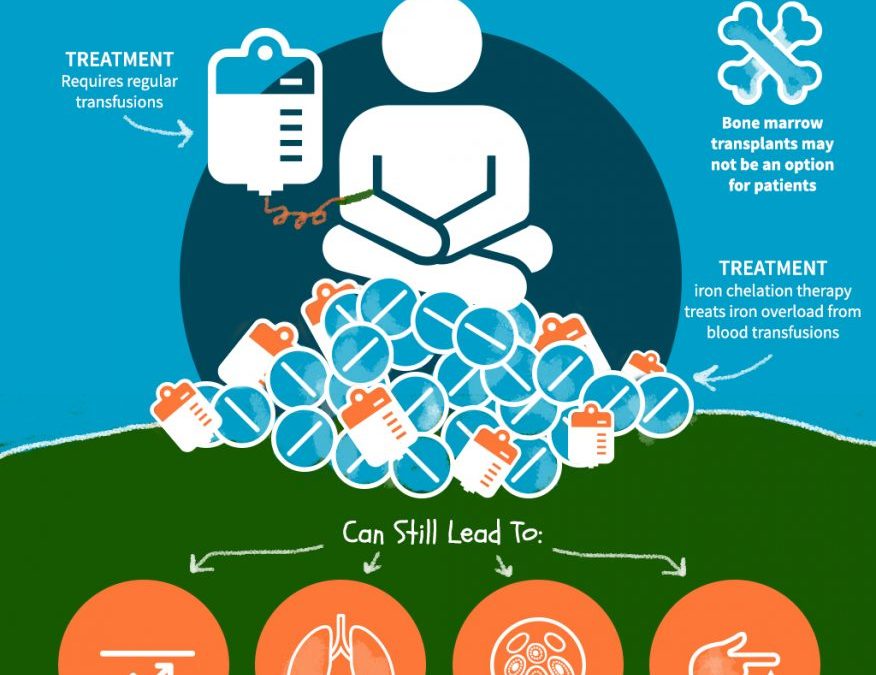
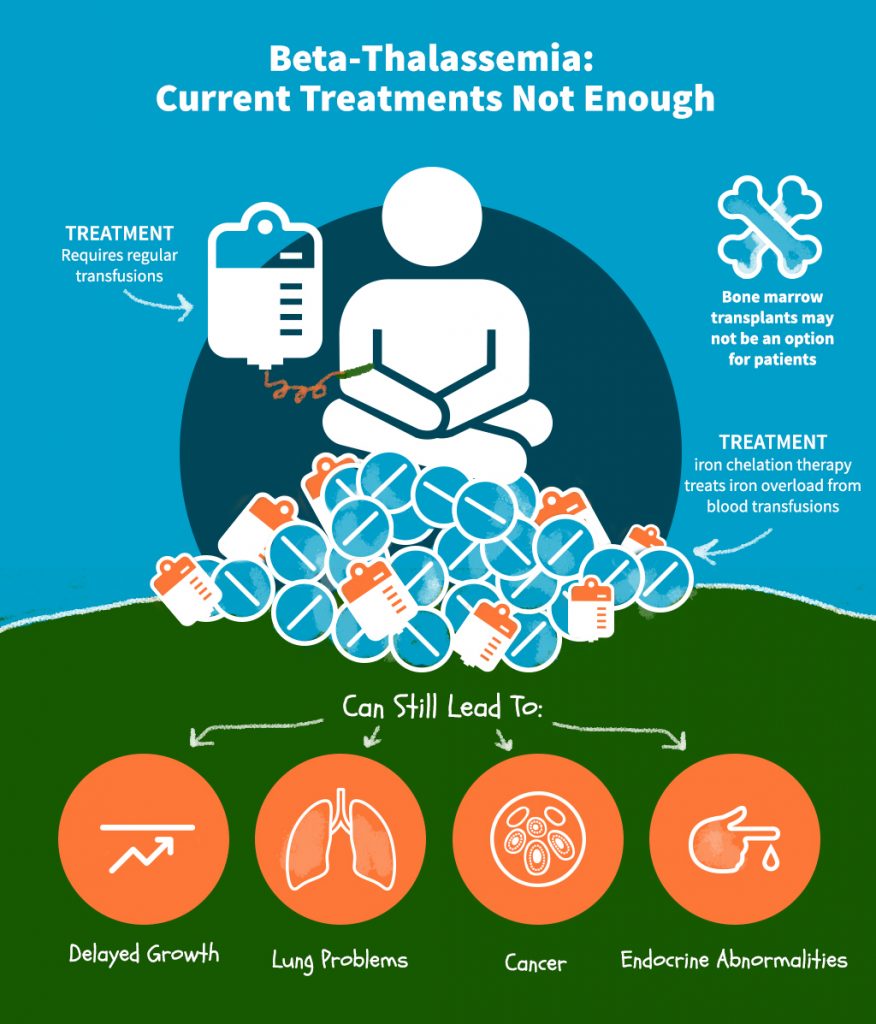
Conventional approaches for treatment
Conventional therapies for beta thalassemia major include lifelong blood transfusion, iron chelation and splenectomy. These approaches focus on palliative management of the disease as well as side effects that result from treatment. Since this involves tremendous cost for healthcare systems and patients, alternative measures are being explored, namely, pharmaceutical Induction of γ-globin, allogenic transplantation, and gene therapy.
Bringing back Fetal Hemoglobin
Pharmaceutical induction of γ-globin is done with the intention of enabling formation of fetal hemoglobin. Epigenetic mechanisms prevent the expression of γ-globin and hence prevent formation of fetal hemoglobin. Removal of this regulation is expected to enable sufficient expression of fetal hemoglobin to compensate lack of functional adult hemoglobin.
A number of drugs have been tested for this purpose – 5-azacytidine, arginine butyrate and hydroxyurea (HU). Currently, HU is the only approved drug for the purpose of γ-globin induction. These drugs are administered in combination with erythropoietin (EPO), which has proliferative and anti-apoptotic properties. The mechanism of action of other compounds is not fully understood and further research is in progress.
Allogenic Stem Cell Transplantation
While conventional therapies are focused on improving longevity and quality of life, they do not provide a permanent cure to Beta thalassemia major. With a view to obtain definitive cure for the disease the first allogenic stem cell transplantation was performed in 1982. Since then, several treatment centers all over the world have performed further research to identify risk factors – disease severity, age at the time of transplantation, stem cell source, histocompatibility and conditioning regimen – and diversify procedures to treat more patients and improve overall thalassemia free survival subsequent to treatment.
One of the main barriers to allogenic stem cell transplantation is the limited availability of donors. In case a sibling with matching HLA is not available, the likelihood of finding an unrelated donor with HLA compatibility varies according to the ethnicity of the patient. Cord Blood (CB) stem cells have been used with varying degrees of success. However, more clinical trials are required.
Gene Therapy
The endeavor to treat thalassemia by gene therapy began at UCLA in 1978. However, the researchers ecountered several difficulties and initial experiments were not successful. After overcoming several technical challenges, gaining understanding of gene regulation and properties of stem cells and development of safer vectors, i.e., lentiviral vectors which lack elements that can activate oncogenes; gene therapy was performed on an 18 year old transfusion dependent thalassemia major patient in 2012. Subsequent to treatment, the patient had normal hemoglobin levels and became transfusion independent.
This provided the impetus for more clinical trials for gene therapy to obtain a better understanding of factors underlying the success of the therapy. Further studies are still required before gene therapy is considered a first line treatment for Beta-thalassemia major.
Reducing the Burden – Genetic Counseling
Since thalassemia is not sex-linked and affects only individuals homozygous for the trait, and carriers are asymptomatic, the World Health Organization (WHO) recommends screening and genetic counseling to be included as part of healthcare systems management of hemoglobinopathies including thalassemia, with a view to reduce the number of new patients born in countries with high incidence of the disease. Greece and Cyprus, with significantly high proportion of carriers of thalassemia, introduced genetic counseling in their healthcare systems and initiated awareness programmes. This resulted in a significant decline of thalassemia cases over a period of 3 decades, which signifies the success of the programme.
Further Reading
- Edouard de Dreuzy , Kanit Bhukhai , Philippe Leboulch, Emmanuel Payen (2016) Current and future alternative therapies for beta-thalassemia major. Biomedical journal 39: 24 -3 8
- J. Weatherall (2001) Phenotype—genotype relationships in monogenic disease: lessons from the thalassaemias Nature Reviews Genetics2:245–255
- Kyrri AR, Kalogerou E, Loizidou D, Ioannou C, Makariou C, Kythreotis L, Phylactides M, Kountouris P, Angastiniotis M, Modell B, Kleanthous M.(2013)The changing epidemiology of β-thalassemia in the Greek-Cypriot population. Hemoglobin. 2013;37(5):435-43. doi: 10.3109/03630269.2013.801851.
- Loukopoulos, D. (2011). Haemoglobinopathies in Greece: prevention programme over the past 35 years. The Indian Journal of Medical Research, 134(4), 572–576.
- WHO Bulletin : http://www.who.int/bulletin/volumes/86/6/06-036673/en/
Related Products
HBB, Monoclonal Antibody (#MBS853525)
HBB, Polyclonal Antibody (#MBS9201956)
HBB, Monoclonal Antibody (#MBS415086)
HBB, Monoclonal Antibody (#MBS854652)
HBB, Polyclonal Antibody (#MBS7600001)
HBB, Polyclonal Antibody (#MBS9132742)
HBB, Monoclonal Antibody (#MBS415221)
HBB / Hemoglobin Beta, Monoclonal Antibody (#MBS246472)
Hemoglobin, Polyclonal Antibody (#MBS171027)
Hemoglobin, Polyclonal Antibody (#MBS171045)
Hemoglobin, Recombinant Protein (#MBS7111107)
Hemoglobin Beta, ELISA Kit (#MBS761707)




